리눅스에서 FASTQ파일 다루기
🟦 fastq파일이란?
fastq파일이란 시퀀싱 결과물로서, 한 시퀀스(시퀀싱 된 read) 정보와 퀄리티 정보를 같이 가지고 있으며, 한 sequence당 총 4줄의 데이터를 가지고 있다. fastq파일의 예시를 보자.
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+SEQ_ID
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65@SEQ_ID
- 시퀀싱 기계에서 지정한 read의 ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
- read의 서열
+SEQ_ID
- 동일한 ID(혹은 다른 아이디지만 SEQ_ID와 일치하는 다른 아이디 들)가 위치
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
- 서열의 각 nt의 퀄리티(Q) 점수
- 퀄리티 점수는 아스키코드의 순서를 따르며 순서는 아래와 같다.
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
이를 보면 서열의 첫 번째 nt인 G는! 에 해당하는 퀄리티 점수를 가진다.! 는 0 값을 가진다. 각 기호와 Q값은 아래 표로 나타내었다.
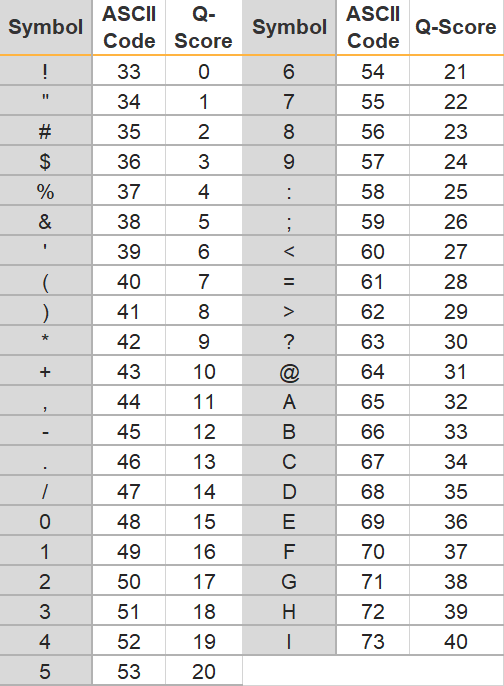
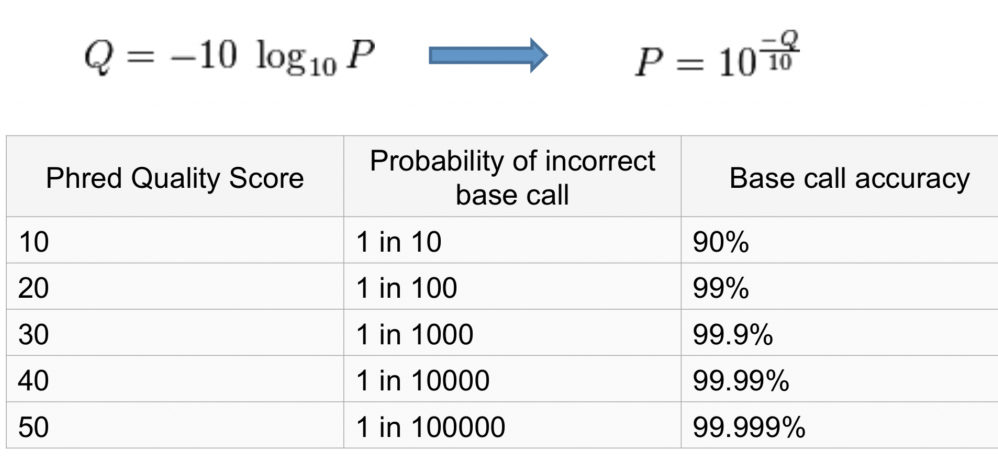
각 Q값은 오류로 인식되었을 확률 값을 기반으로 측정된다. 만약 Q값이 20이라면, 오류가 날 확률은 10^-2로, 100번 중 한번 꼴로 오류가 나서 잘못 인식되었 가능성이 있다는 것이다. 그러므로 Q값이 높을수록 오류확률이 낮고, 시퀀싱 된 서열의 정확가 높다고 할 수 있다.
🟦 리눅스에서 다루기
1. fastq파일 들여다 보기
less file.fastq파일 보기
tail -n +900 file.fastq | head-15900번째 줄에서 top 15줄만 보기
2. fastq 파일 안에 있는 sequence read 수 세기
grep -c "^>" file.fastqfile.fasta파일에서 read 수 세기
grep -c "^+$" file.fastqfile.fasta파일에서 quality score lines 세기
zcat file.fastq.gz | echo "$((wc -l / 4))"줄 개수 %4 = 파일 안의 sequence 수
zcat file.fastq.gz | wc -l | awk '{print $1 / 4}'awk 함수 이용하기
for fname in *.gz; do
echo "Processing $fname"
echo "..$fname has `zcat $fname | wc -l | awk '{print $1 / 4}'` sequences"
done여러 fastq 파일의 서열 개수 확인하기
3. 현재 디렉터리에서 fastq 파일 목록 보기
wc -l *.fastq.gz현재 디렉터리에서. fastq.gz인 파일 목록 보기
wc -l *.fastq.gz > list.txt현재 디렉토리에서 .fastq.gz인 파일 목록 저장하기
4. 전체 분석 샘플 개수 확인하기
일루미나 사의 시퀀서는 총 두 가지 서열로 forward와 reverse 서열리다. 그러므로 시퀀싱된 결과파일은 시퀀싱을 돌린 샘플 수 X 2 이다. 역으로 결과물 /2 를 하면 샘플의 수를 알 수 있다
echo "$((`ls -l | grep ^- | wc -l`/2))" fastq파일 개수로 샘플 수 추정하기 (file 수 /2)
5. 각 서열 pair인지 확인하기
zcat file_R1.fastq.gz | head
zcat file_R2.fastq.gz | headzcat file_R1.fastq.gz | wc -l
zcat file_R2.fastq.gz | wc -l
6. 원하는 염기 서열패턴을 가신 read 찾기
grep AACTGCTA -B 1 file_R1.fastq.gzfile_R1. fastq.gz에서 AACTGCTA패턴을 가진 서열과, 그 서열 전(before) 부분의 한 줄을 같이 출력해라
grep '[TGCA]\{2,\}' file_R1.fastq.gzTGCA가 두 번 이상 반복되는 경우 출력
grep '[A-Z]\+[1-9][0-9]' file_R1.fastq.gz대문자 한번, 1-9사이 숫자, 0-9 사이 숫자가 나오는 패턴 출력
🟦 참고
- https://en.m.wikipedia.org/wiki/FASTQ_format
- https://wikis.utexas.edu/display/CoreNGSTools/Working+with+FASTQ+files
- https://bioboot.github.io/web-2016/class-material/day3-fastq-unix-practice.html#
- 빅데이터&인공지능 with 생물정보학(양우진/아이콕스)
- https://learn.gencore.bio.nyu.edu/ngs-file-formats/quality-scores/