[MultiQC] conda를 사용한 간단한 설치 및 실행

🟧 NGS data workflow
보통 연구실에서는 시퀀서 가격이 비싸다 보니, 회사에 시퀀싱을 맞기게 됩니다. 이때 결과물로 raw data와 NGS report를 전송해 주는데, 이 안에 기본적인 QC정보를 담고 있습니다.
그러나 이 보고서는 raw data가 쓸 만 한지 평가해 주진 않습니다. 그러므로 NGS데이터 분석에서 가장 중요한 것은 Raw data가 신뢰할 만한 것인지 판단하는 것입니다.

이를 위해 가장 대중적으로 사용되는 프로그램에는 fastQC와 multiQC가 있습니다.
🟧 설치 및 실행하기
multiQC는 fastQC의 보고서를 하나로 합쳐주는 것으로, 실제 QC분석은 fastQC에서 구동됩니다.
- fastqc 홈페이지: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- multiqc 홈페이지: https://multiqc.info/docs/#installing-multiqc
다운로드 방법에는 여러 가지가 있지만, 이 중에서 conda를 사용해 보겠습니다.
# 1. python > 3.8
conda create --name py3.11 python=3.11
conda activate py3.11
# 2. install fastqc
conda install fastqc
# 3. install multiqc
conda install multiqc
# 4. run fastqc
fastqc -o results/ *.fastq.gz
# 5. run multiqc
multiqc results/
🟧 예제 보고서 확인하기
MultiQC 홈페이지에서 예제 보고서를 확인할 수 있다.
- RNAseq: https://multiqc.info/example-reports/rna-seq/
- Whole-Genome Seq: https://multiqc.info/example-reports/whole-genome-seq/
각 데이터에 따라 평가되는 속성이 일부 다르다. 아래 보고서는 fungi ITS1의 paired-end 서열 fastq파일의 분석 보고서이다.
General Statistics
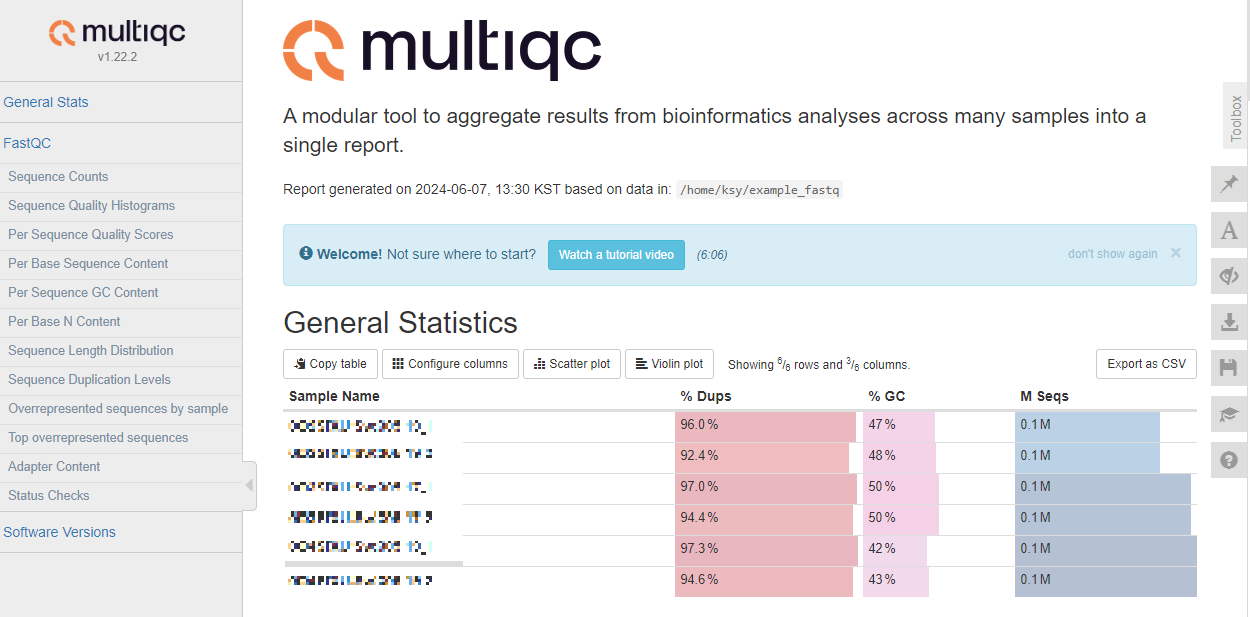
가장 위에 있는 General Statistics를 통해 주요 통계정보를 한눈에 볼 수 있다.
- Configure columns에서 보여주는 옵션을 추가할 수 있다.
FastQC
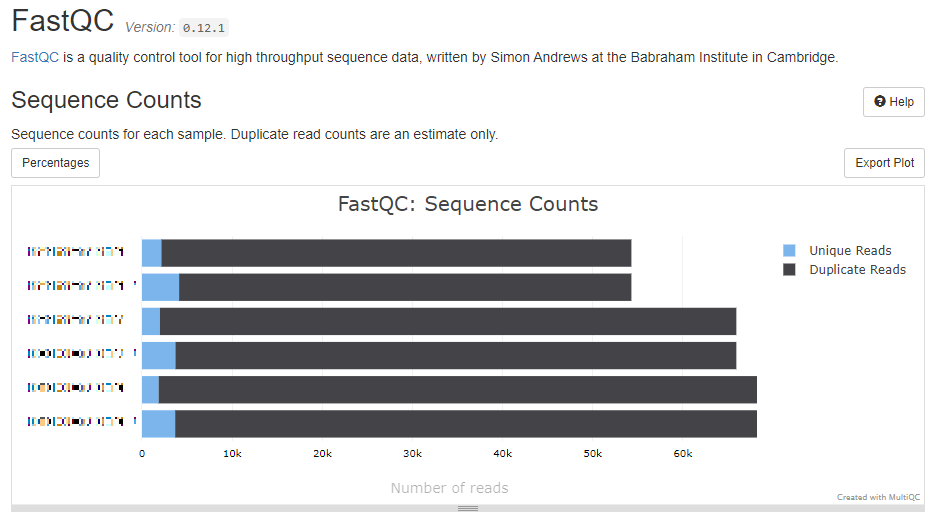
하나의 샘플에 몇 개의 read가 존재하는지 보여준다. 이때 중복되는 서열은 “Duplicate Reads”로 표시된다.
그러나 분석을 위해 모두 50dp로 잘려서 비교된다. 그러니 read수만 확인하고 넘어가자.
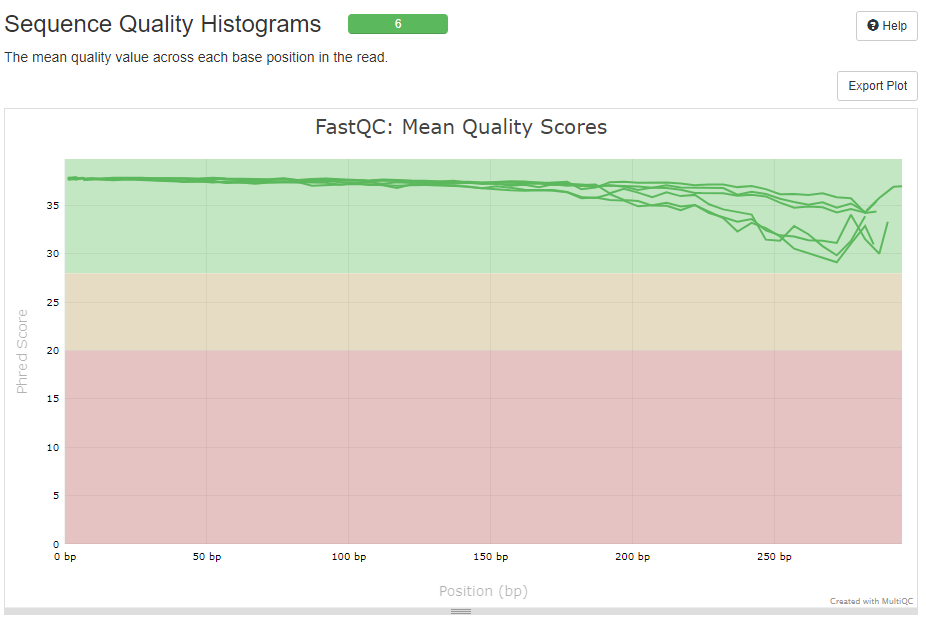
Sequence Quality Histograms

각 염기 위치에 따른 평균 품질 점수를 보여준다. 마우스를 가져가 대면 해당 샘플의 이름을 알 수 있다.
- Green = good quality
- Oragne = resonable quality
- Red = poor quality
전체 6개 중 하나의 샘플만 경고 수준이며, 나머지는 fail 수준임을 알 수 있다. 0bp 부분을 마우스로 박스 모양을 만들어서 확대해 보자.
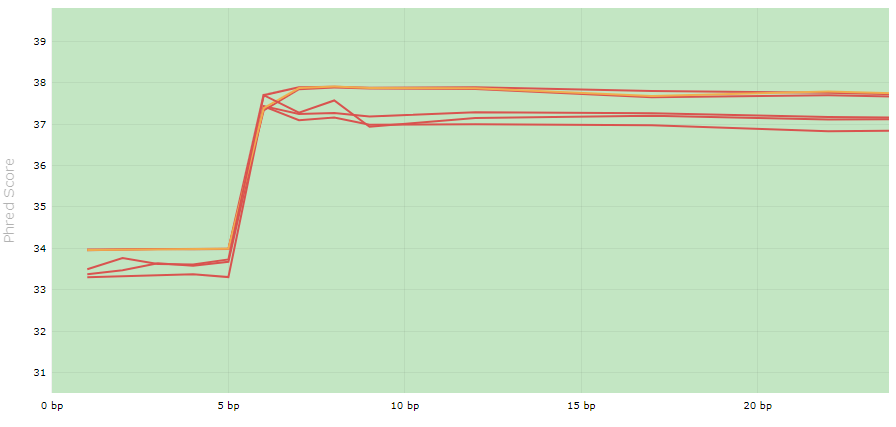
앞부분의 퀄리티가 하락하는 것으로 보아 잘리지 않은 primer의 위치로 보인다.
Per Sequence Quality Scores
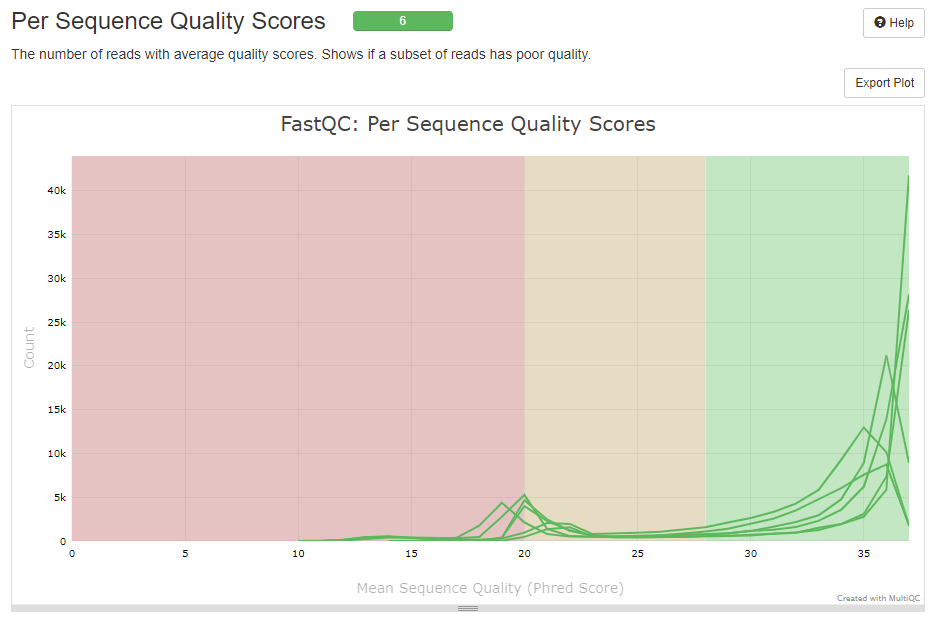
각 샘플에서 품질 점수에 따른 빈도를 보여준다.
- Warning = 평균 QC < 27
- Failure = 평균 QC < 20
Per Base Sequence Content

염기 위치에 따라 4개의 DNA염기 call 비율을 나타난다.
- warning = A와 T 또는 G와 C의 차이가 10%보다 클 경우 경고
- failure = A와 T, 또는 G와 C의 차이가 20%보다 크면 quality fail
Per Sequence GC Content

각 read에 따른 GC 비율의 평균값을 나타낸다. 일반적으로 GC 양은 정규분포를 따른다.
- 그러나 6개의 샘플은 모두 정규 분포를 따르지 않음
Per Base N Content

N의 개수를 나타낸다. N은 시퀀서가 품질에 대한 확신이 없을 때 나타나는 결괏값이다.
- warning = 5 % 이상의 N
- failure = 20 % 이상의 N
Sequence Length Distribution

서열의 길이를 나타낸다.
- Miseq을 사용하여 시퀀싱 된 샘플로 대부분이 300bp의 길이를 가진다.
Sequence Duplication Levels

중복된 서열의 비율을 나타낸다. 75bp이상인 read는 모두 50bp로 잘린 후 분석된다.
- warning = 중복되지 않는 sequence가 전체의 20 % 이상을 차지
- failure = 중복되지 않는 sequence가 전체의 50 % 이상을 차지
중복된 서열이 많을수록 PCR에 의한 Bias의 결과로 추정된다. 그러나 Amplicon데이터 특성상 마이크로바이옴 결과에서는 중복 값이 많을 수 있다.
Overrepresented sequences by sample

- 0.1% 이상의 중복된 서열의 비율을 나타낸다. 길이가 길면 50bp로 잘린 후 분석된다.
Top overrepresented sequences

최소 0.1% 이상의 서열의 복제 수준을 나타냄
Adapter Content

기존에 알려진 adapter서열을 검색하여 샘플 내에 얼마나 존재하는지 확인한다. 좋은 데이터는 adapter가 존재하지 않는다.
- Warning = 5% > adapter
- Failure = 10 % > adapter
Status Checks

전체적인 결과가 정상인지 fail인지 알려준다.
After quality filtering
위 6개 파일은 결과적으로 좋지 못한 퀄리티를 나타낸다. 그렇다면 Adapter를 자르고 QC를 평균 30 이상인 read만 남긴다면?
확실히 파일 샘플의 평균 quality는 좋아졌다.

- Dulication과 Overrepresented는 Amplicon데이터를 50 bp로 잘라서 확인하기 때문에 중복이 많은 건 어쩔 수 없는 결과이다.
- Per base sequence와 Adapter Content는 좋아졌지만, 오히려 Per Tile Sequence Quality는 안 좋아졌다.
🟧 참고
- https://www.youtube.com/watch?v=1YkkRGrfw5k
- https://m.blog.naver.com/pickyu2/222980753788