Bioinformatics/기타
[R] RNA-seq의 log2 DEG 데이터로 heatmap 그리기
김해김씨99대손
2022. 9. 21. 12:46
자료 설명
▶ 자료 출처 : https://www.edwith.org/ptnr/kobic/ 의 <전사체 데이터 분석> 강의
▶ 샘플 정보
- 3반복 실험
- CDA Knockout 과 Control
- 총 6 sample
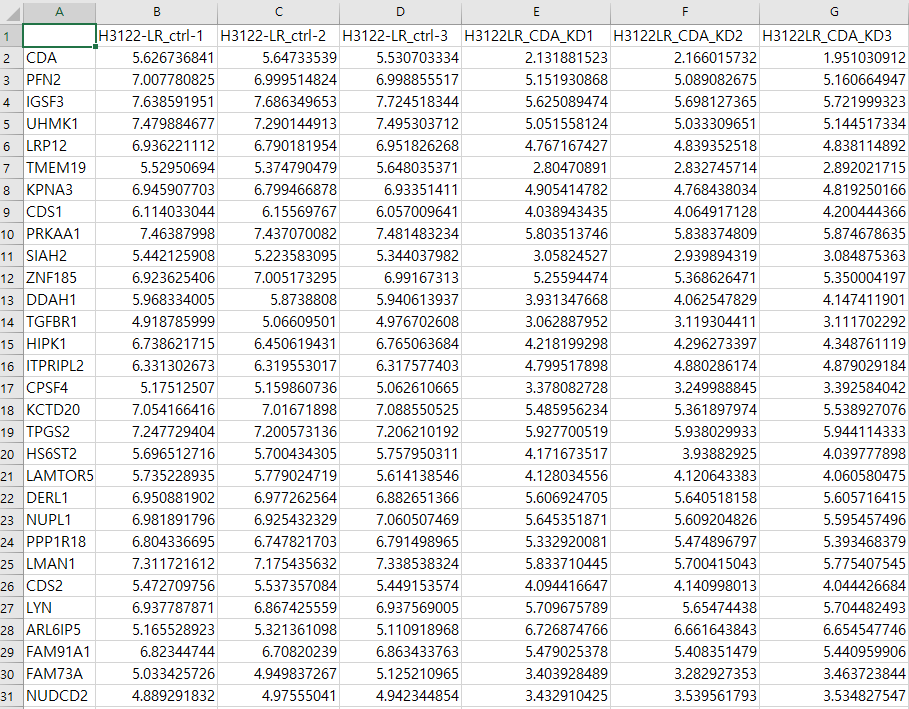
▶ DEG 파일 정보 (log2 read count table)
- log2로 치환한 값
- raw name은 gene ID, column name은 sample ID
R에서 heatmap 그리기
1. 파일 읽어 오기
DEG <- read.table("./edgeR_DEG_expression.txt", sep = '\t', header = T, row.names = 1)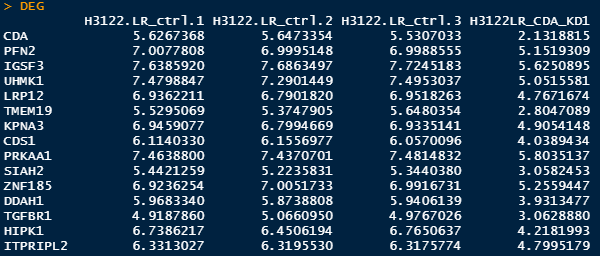
1. heatmap 그리기
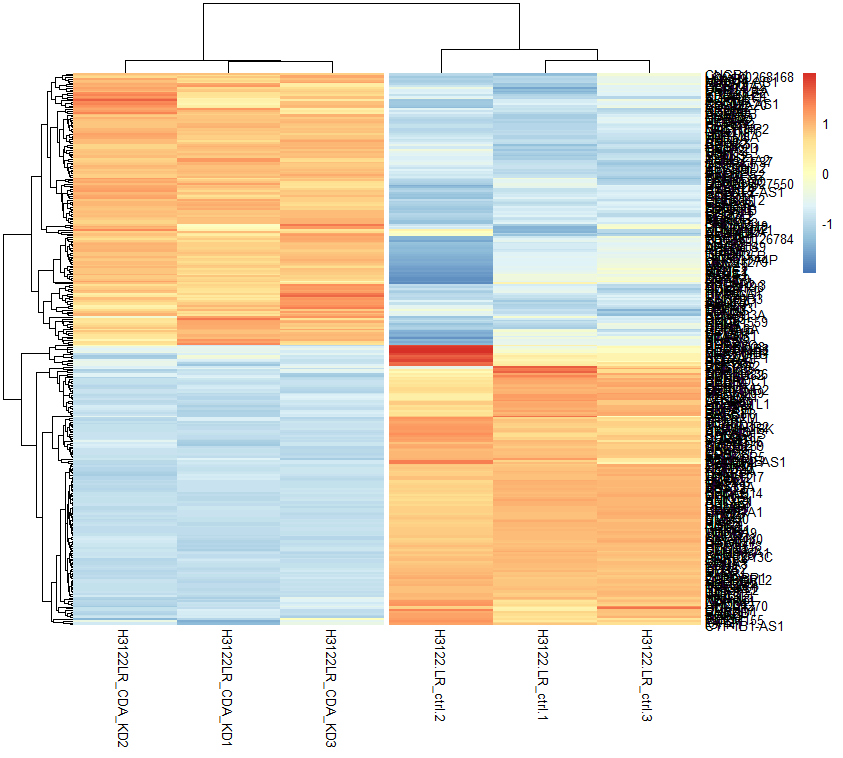
library(pheatmap)
pheatmap(DEG,
cutree_cols = 2, # sample 을 2그룹으로 나누어 흰 선을 그어준다
scale = 'row' # row 별로 발현량의 차이를 잘 보기 위해 nomarization 해준다
)
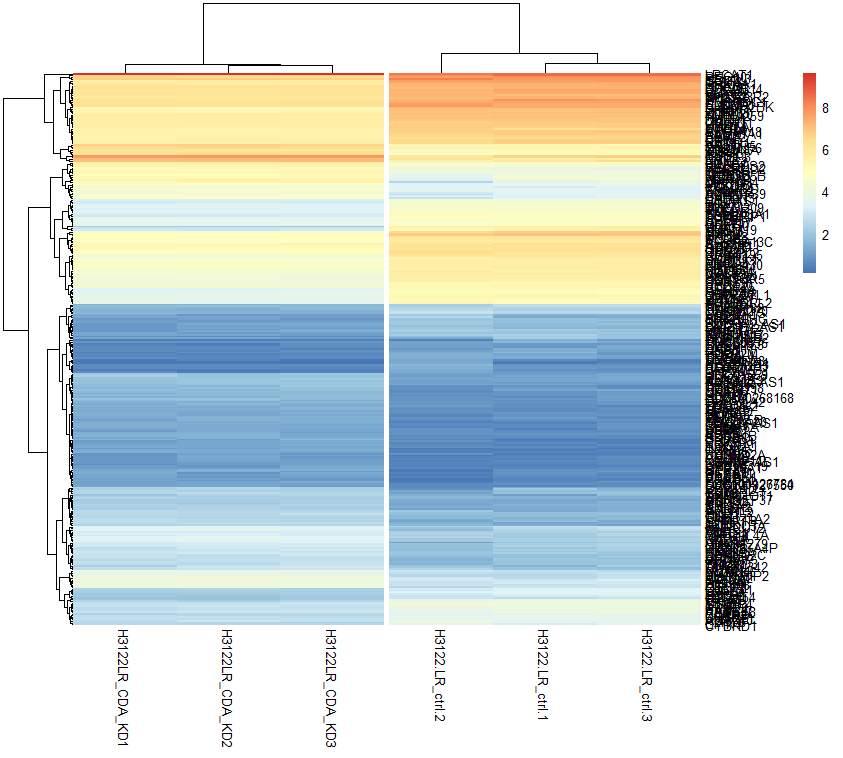
만약 scale = 'row' 를 추가하지 않는다면 아래와 같이 나오게 된다
만약 아래와 같이 sample정보가 추가된 축을 추가하고 싶다면?
Colors = colorRampPalette(c("blue", "white", "red"))(100) #heatmap 색 조정
# bluw~ white~ red 사이를 100개의 구역으로 나누어 색을 표현한다는 뜻
ann_col <- data.frame(type = as.factor(c(rep("CDA", 3), rep("Knock_down", 3)))) # sample의 type 나타내기
rownames(ann_col) <- c("H3122.LR_ctrl.1","H3122.LR_ctrl.2","H3122.LR_ctrl.3" ,
"H3122LR_CDA_KD1", "H3122LR_CDA_KD2", "H3122LR_CDA_KD3") # rownames을 sample name으로 지정해 주어야 한다
ann_color <- list(type = c(CDA = "purple",Knock_down = "yellow")) # annotation 의 색을 정해준다
### Run pheatmap
pheatmap(DEG,
color = Colors,
annotation_colors = ann_color,
border_color=NA,
scale="row",
show_rownames=F,
annotation_col = ann_col,
#annotation_row = ann_row,
#cluster_rows = F, cluster_cols = F, # sample 정렬 X -> dendrogtam X
#gaps_row = 100,
#gaps_col = 3,
legend = T)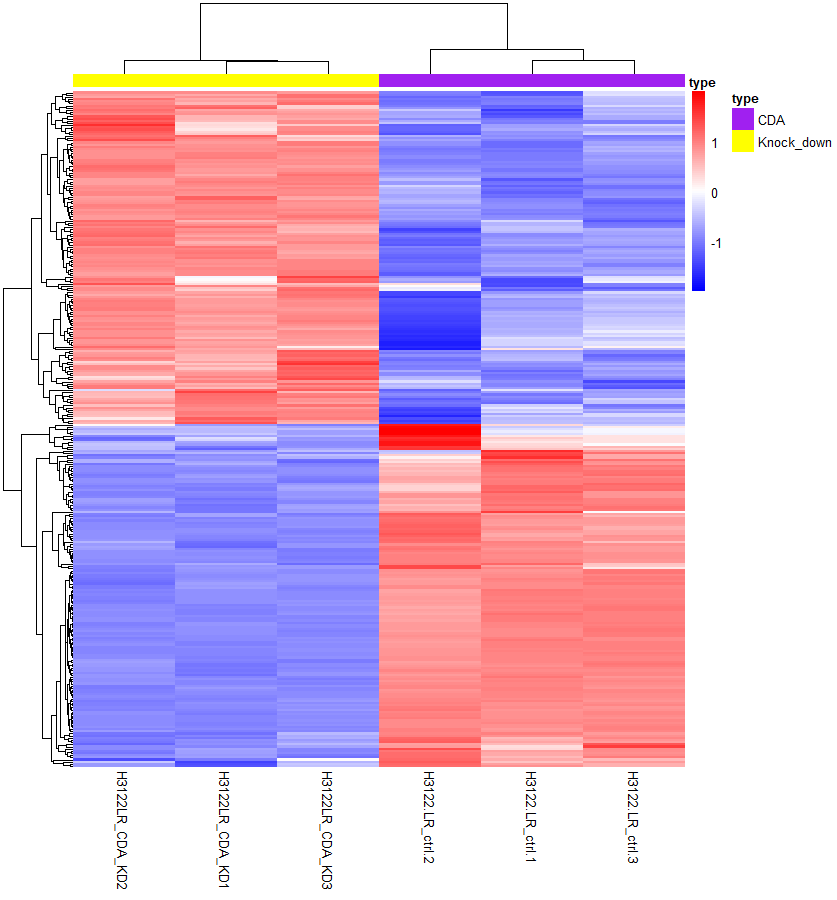
Reference
- https://hbctraining.github.io/Intro-to-R-with-DGE/lessons/B1_DGE_visualizing_results.html
반응형