[MEGA11] 설치 및 기본 예제 파일로 계통수 그려보기
수정 : 2023-04-16

프로그램에 관한 사용법임으로 분류 알고리즘에 대한 설명은 생략하겠습니다.
1. 다운로드 및 설치
- MEGA 다운로드 링크 : https://www.megasoftware.net/


MobaXterm_Installer_v22.1 압축 풀기

MobaXterm_Installer_v22.1 더블클릭 -> 모두 Yes -> 설치 완료
2. 예제 파일로 계통수 그리기
1) FASTA format으로 저장하기
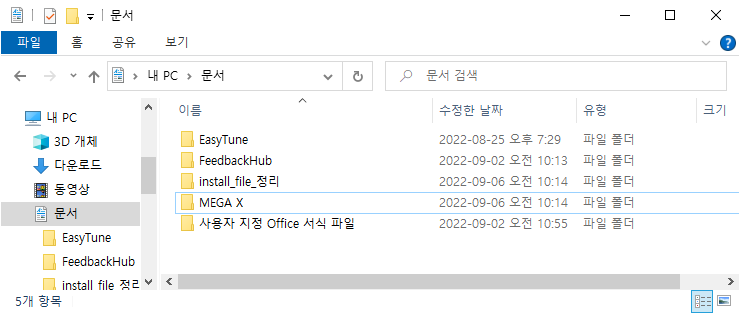
"내 PC\문서"를 가면 "MEGA X" 폴더가 만들어진 것을 볼 수 있습니다.
그 안의 "Examples"폴더에 들어가면 MEGA프로그램 사용법을 익히기 위해 추가적으로 다운된 샘플들의 fasta파일들을 볼 수 있습니다.
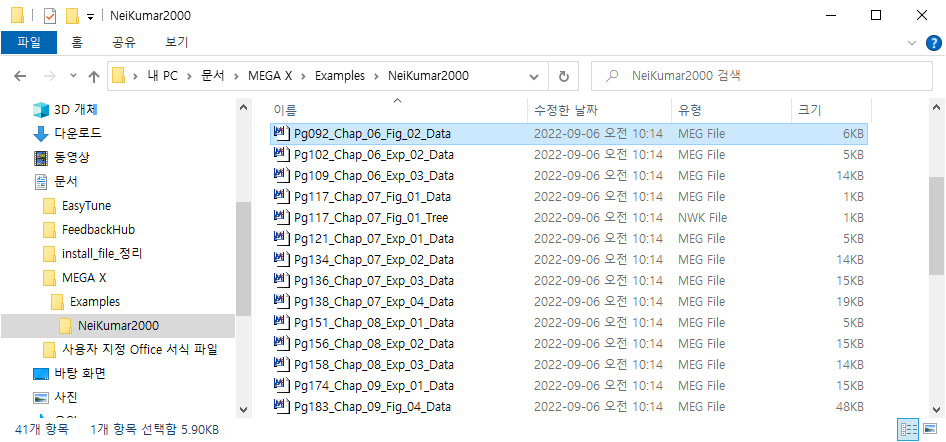
"MEGA X\Example\NeiKumar2000" 폴더에 들어가 Pg092_Chap_06_Fig_02_Data.meg 파일을 더블클릭 해보자.
meg파일은 MEGA에서 사용되는 저장 형식으로 MEGA프로그램 안에 파일 하나가 추가된 것을 볼 수 있다.
왼쪽 네모 아이콘(TA)을 클릭하면 MEGA프로그램으로 import된 파일을 볼 수 있다
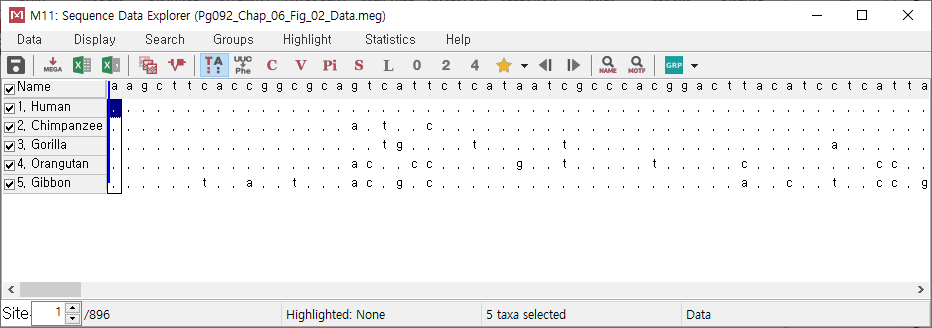
위 파일을 살펴보자
Human 서열은 모두 '.'으로 표시되어있고
다른 영장류 에는 '.'과 DNA를 구성하는 acgt가 뜨문뜨문 표시되어 있다
이는 맨 위에 (Name) 줄의 서열이 human에 해당하는 서열이고, 나머지는 그 서열과 다른 염기만 표시해 둔 서열임을 알 수 있다. 하지만 이것은 우리가 아는 fasta파일이 아님으로 계통수를 그리기 위해 fasta format으로 저장해주어야 한다.
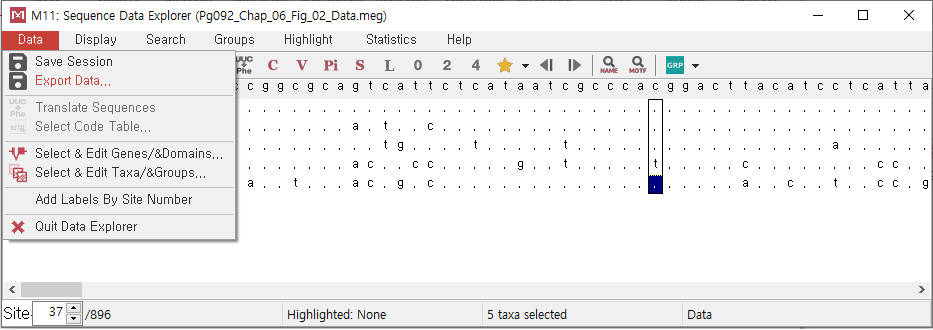
"Data" -> "Export Data" 클릭
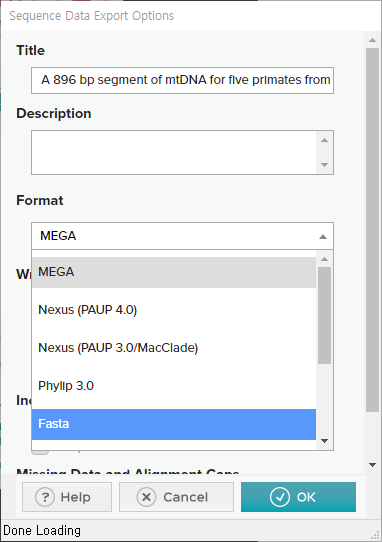
format을 "Fasta"로 바꾸어 주고 -> "OK"
이후 "저장" 클릭
이후 결과를 볼 거냐고 물어보는 창이 뜨는데 한번 보면("YES")
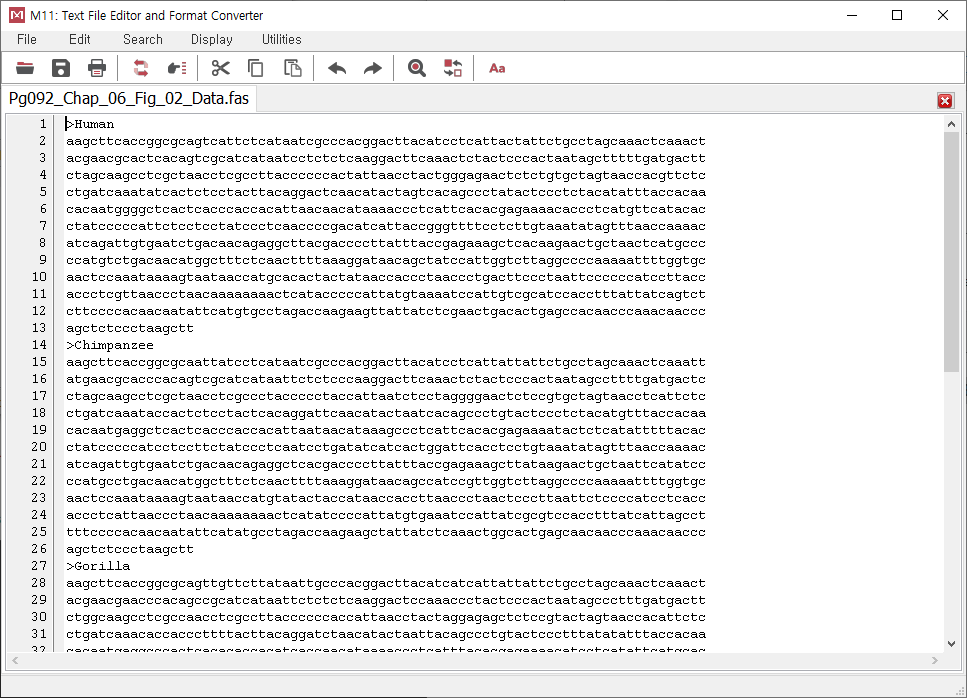
위와 같이 저장된 fasta format으로 저장된 것을 볼 수 있다
2) Alignment 하기
만들어진 "Pg092_Chap_06_Fig_02_Data.fas"을 더블클릭 한다
이후 뜨는 알림 창의 "Align"을 클릭한다.
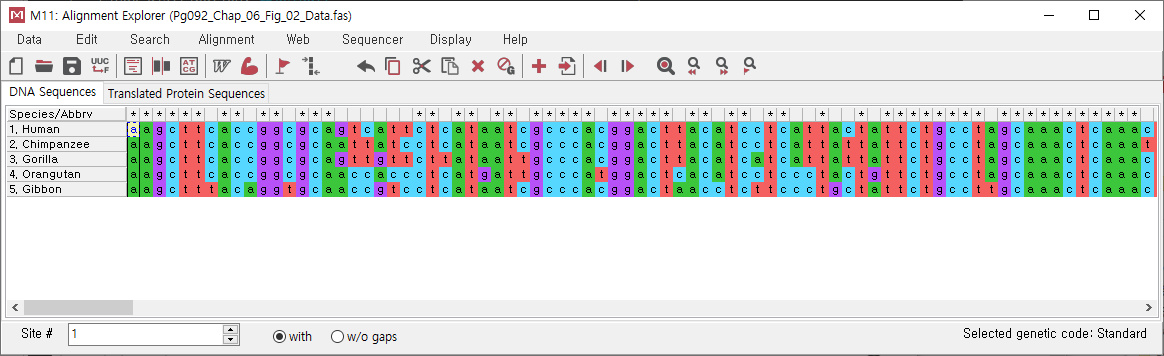
총 896 개의 염기가 정렬된 것을 볼 수 있다.
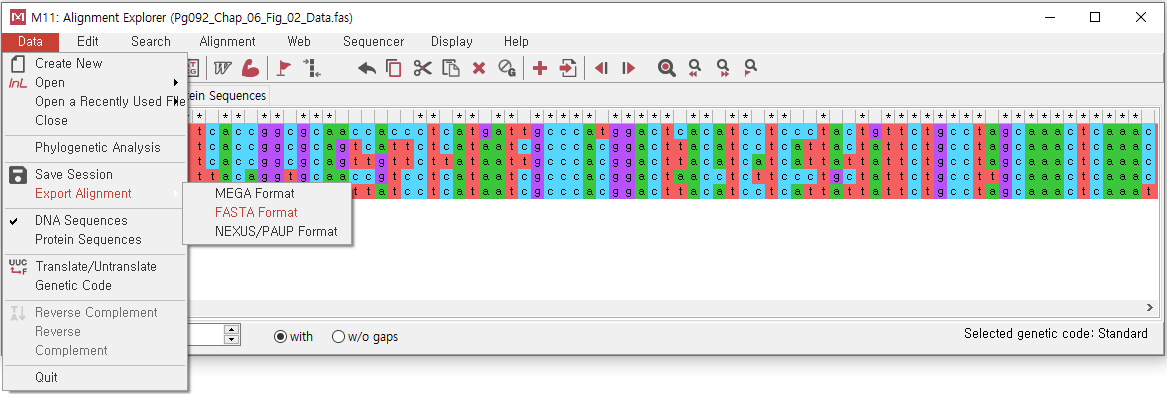
"Data" > "Export Alignment" > "FASTA Format" 클릭
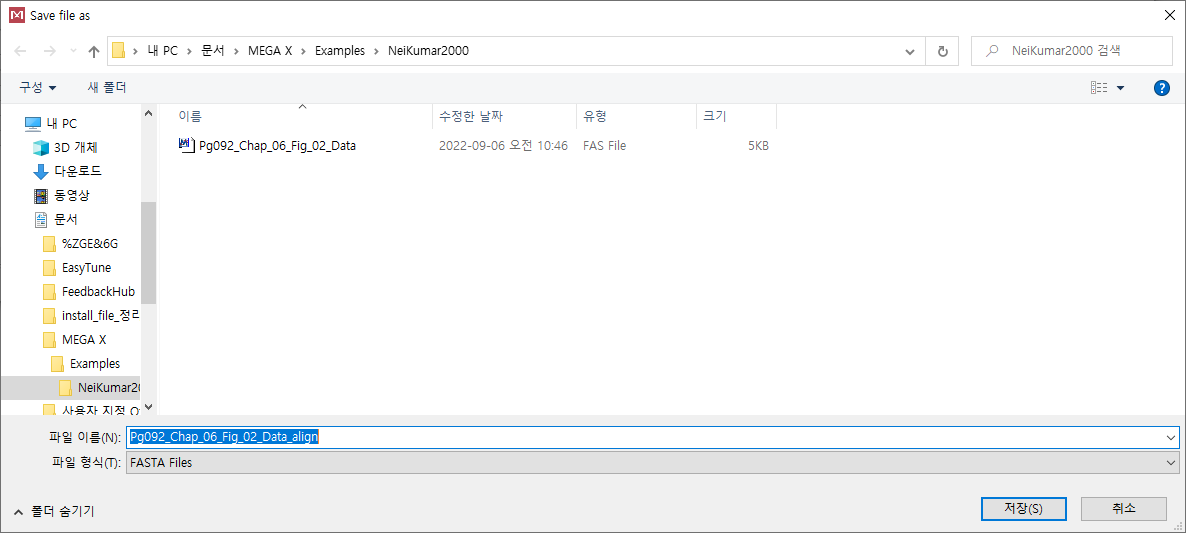
이름을 "Pg092_Chap_06_Fig_02_Data_align"으로 수정한 후 저장해 준다
3) Phylogeny Tree 그리기
이번에는 Mega에서 파일을 열어보자
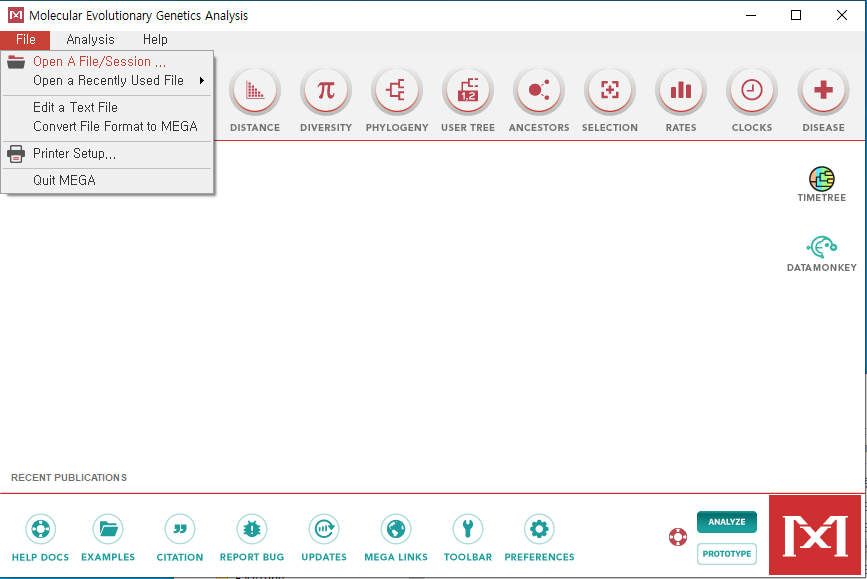
"File" > "Open A File/Session" 클릭 -> "Pg092_Chap_06_Fig_02_Data_align" 클릭
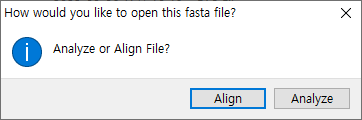
이미 Align을 했으므로 "Analyze"를 클릭해 준다
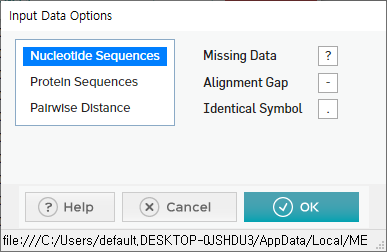
DNA이니까 Nucleotide Sequence선택 -> "OK"
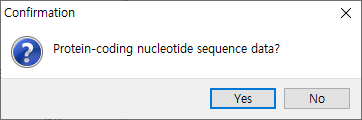
이후 알림 창에서 DNA -> Protein 으로 바꾼 후에 데이터도 비교해 볼 거냐 물어본다
그러나 지금은 예제 데이터이니 DNA서열만 관찰해 보자
"No" 클릭
"PHYLOGENY" 아이콘에 마우스를 대면 위와 같은 옵션을 볼 수 있다
일단 "Neighbor-Joining Tree"로 계통수를 그려보자
"Yes"
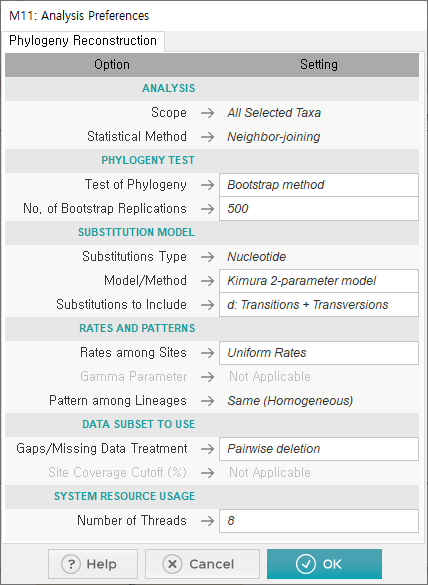
추가적인 옵션을 설정해 보자.
Phylogeny test를 "bootstrap 500"으로 바꾸고 -> model을 "kimura"로 바꾸고 -> "OK"
이제 bootstrap이 끝나면 아래와 같은 계통수를 얻을 수 있다.
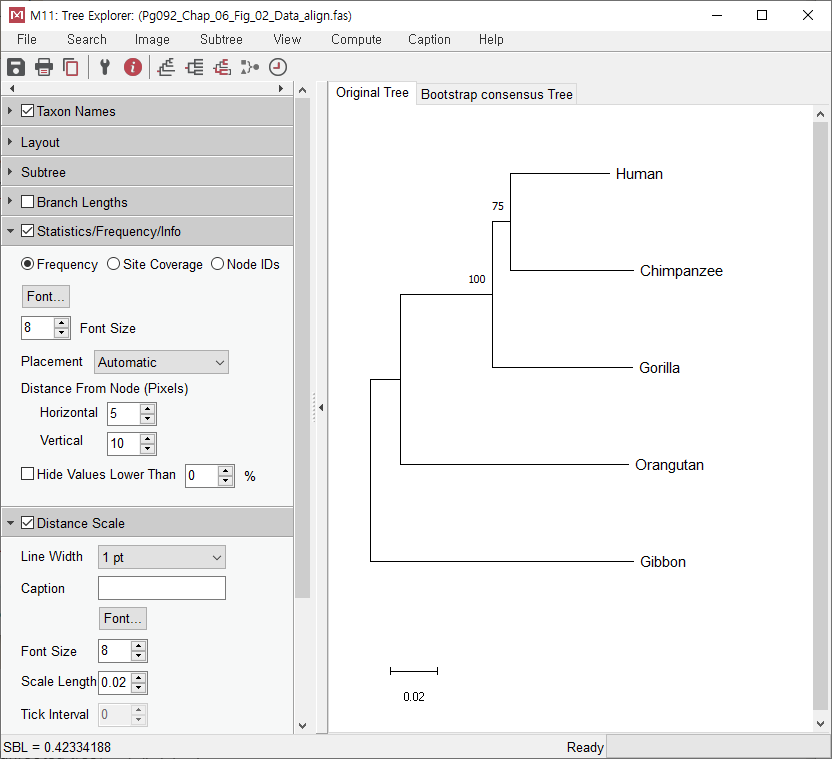
이제 생성된 계통수를 해석해 보자
Q. 100, 75는 무엇을 뜻하는가?
bootstrap 500번 돌렸을 때 human과 chimpanzee는 전체 500번 중에 75%가 같은 그룹에 걸렸고 나머지는 100%의 확률로 같은 그룹으로 묶였다
Q.0.02는 무엇을 뜻하는가?
각 node마다 distance가 얼마나 떨어져 있는지를 나타낸다. 0.02를 나타내는 막대의 길이와 Gibbon과 Orangutan의 거리를 측정해 보았을 때 distance는 약 4.5*0.02 = 0.09로 볼 수 있다.
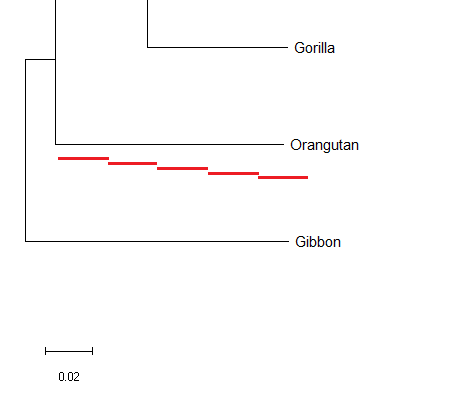
추가적으로 다른 형태의 계통수도 그려볼 수 있다.
위 옵션으로 tree의 구조를 변형하여 볼 수 있음
그다음 "Maximum parsimony"로 계통수를 그려보자
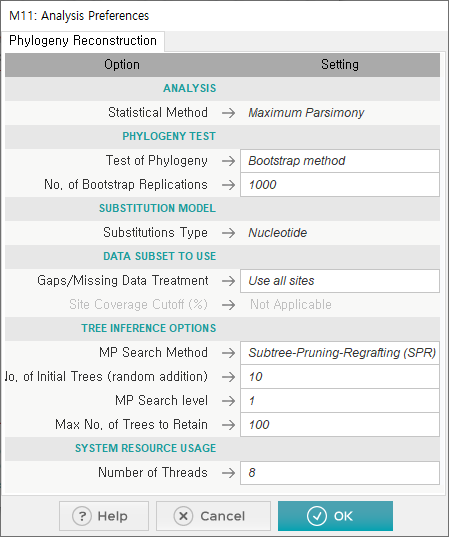
bootstrap은 1000으로 설정하고 느긋하게 기다려보자.. 시간이 꽤 걸린다.
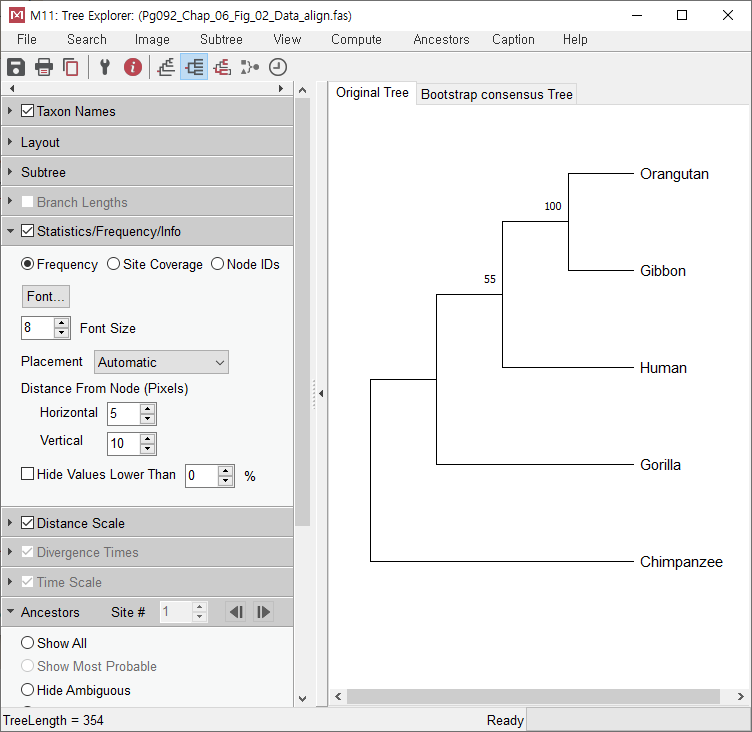
Maximum parsimony는 아까와 달리 distance가 나타나지 않는다. Neighbor-Joining Tree는 각 서열들의 차이를 distance로 계산하여 계통수를 구하지만 Maximum parsimony는 distance가 아닌 character based방법으로 염기서열 각각 하나씩 비교하는 방법이 기 때문이다.
위 그림에서 100과 55가 뜻하는 것은 Orangutan과 Gibbon이 1000번 중 1000번 다 그룹 지어지고,이 그룹과 human은 1000번 중 550번만 같은 그룹으로 짝지어졌다는 뜻이다.
| 추가적으로 볼만한 글
- [R/ggtree] ggtree패키지를 이용해 phyloseq데이터의 계통수 그려보기
Q. 만약 내가 가진 샘플이 alignment 했을 때 gap이 너무 많이 나온다면?
Gap이 생기는 이유는 missing data일 수도 있고 Indel, Structural Variation(SV) 일 수 있다. Missing Data 라면 잘라내면 그만이지만 Indel, Structural Variation(SV)의 경우 진화적 현상일 수도 있으니 잘라내기 어렵다
MEGA에서는 pairwise의 경우 compelete deletion, MP의 경우 partial deletion 옵션으로 indel 계통수에 반영하지 않고 버릴 수 있다
정확성을 위해선 MrBayes* 같은 정확성이 높은 도구에서 nucleotide, amico acid서열과 같이 넣어서 indel여부를 보는 프로그램 사용을 추천한다
* MrBayes : Bayesian inference을 기반으로 하는 계통수 모델. MrBayes uses Markov chain Monte Carlo (MCMC) methods to estimate the posterior distribution of model parameters.
참고 : https://academic.oup.com/sysbio/article/66/5/698/2999315?login=true