[QIIME2 튜토리얼] “Moving Pictures” (2)
2024-04-18 업데이트

이전 편
[QIIME2 튜토리얼] “Moving Pictures” (1)
2024.04.18.업데이트 마이크로바이옴을 공부하면 아마 가장 먼저 배우게 되는 것이 이 QIIME2의 사용법입니다. Moving pictures tutorial을 참고하여 각 단계별로 세세하게 알아봅시다.🙉 분석 데이터 관찰
bio-kcs.tistory.com
- .qzv 포맷 파일은 www.view.qiime2.org에 드래그 앤 드랍하면 볼 수 있다.
- 통계학 적인 개념보단 qiime2 tool에 대한 설명 위주의 글이다.
06. Generate a tree for phylogenetic diversity analyses
계통수를 만들기 위해서는 먼저 서열을 정렬해 주어야 합니다.
이때 QIIME2에서는 align-to-tree-mafft-fasttree 함수를 사용한다. 위 함수는 아래와 같은 단계를 수행합니다.
(1) 서열 정렬(align)을 수행하기 위해 mafft(multiple sequence alignment program) 프로그램을 실행
(2) 정렬하면서, 변이가 너무 많은 부분 삭제하기 위해 표시(masking) 하여 속도를 높인다
(3) 계통수를 만들기 위해 FastTree프로그램 사용, 이는 unrooted tree를 만들어낸다. 마지막 단계에서 계통수의 중간 부위에 root를 추가로 만든다.
qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences rep-seqs.qza \
--o-alignment aligned-rep-seqs.qza \
--o-masked-alignment masked-aligned-rep-seqs.qza \
--o-tree unrooted-tree.qza \
--o-rooted-tree rooted-tree.qza
결과 파일을 봅시다
qiime tools export --input-path aligned-rep-seqs.qza --output-path aligned-rep-seqs/
cd aligned-rep-seqs/
less -S algned-dna-sequences.fasta
아까 보았던 rep-seqs의 dna-sequence.fasta의 파일과 구조가 다른 것이 보이나요?
정렬된 서열은 유사한 부위를 같은 열에 표시하기 위하여 빈 영역을 '----'로 구분되었습니다.
Q. 각 ASV를 그냥 BLAST 하거나, 다른 데이터 베이스와 비교해서 알아낼 수 있는데, 왜 굳이 계통수를 만들어야 하나요??
A. 이유는 diversity 분석을 위해 서열들이 버려지지 않게 하기 위함입니다.
Taxonomy classification 되는 서열만 diversity 분석을 수행할 때, taxonomy Database가 완벽하지 않은 경우 일치하지 않은 서열은 모두 제거됩니다. 그래서 우리는 ASV 단위로 각 서열이 얼마나 유사한지 살펴보는 것입니다.
물론 연구 목적에 따라서 classification을 먼저 하고 diversity를 보는 경우도 있습니다.
07. Alpha and beta diversity analysis
위 단계에서 샘플의 alpha, beta diversity 분석을 수행합니다. 명령어는 간단하지만 분석 방법의 이해가 필요합니다.
QIIME2에서는 core-metrics-phylogenetic 명령어를 사용해 여러 alpha, beta diversity에 해당하는 지수를 모두 계산할 수 있습니다(QIIME의 장점이죠!).
각 다양성 지수를 따로 계산할 수 있지만, 연구에 많이 사용되는 값을 core로 설정해서 한 번에 계산해 주는 함수입니다.
결과물은 아래와 같습니다.
- Alpha diversity :
- Shannon’s diversity index (a quantitative measure of community richness)
- Observed Features (a qualitative measure of community richness)
- Faith’s Phylogenetic Diversity (a qualitiative mea
- sure of community richness that incorporates phylogenetic relationships between the features)
- Evenness (or Pielou’s Evenness; a measure of community evenness)
- Beta diversity
- Jaccard distance (a qualitative measure of community dissimilarity)
- Bray-Curtis distance (a quantitative measure of community dissimilarity)
- unweighted UniFrac distance (a qualitative measure of community dissimilarity that incorporates phylogenetic relationships between the features)
- weighted UniFrac distance (a quantitative measure of community dissimilarity that incorporates phylogenetic relationships between the features)
우리는 유의해야 하는 점이 있습니다. 바로 sampling depth입니다.
qiime diversity core-metrics-phylogenetic \
--i-phylogeny rooted-tree.qza \
--i-table table.qza \
--p-sampling-depth 1103 \
--m-metadata-file sample-metadata.tsv \
--output-dir core-metrics-results
샘플에 따라 depth에 따른 보정을 하지 않는 연구도 있고, 보정을 한다면 어떤 값으로 잡아야 하는지가 중요한 문제입니다.
이 depth는 어떻게 사용될까요?
만약 depth를 1,000으로 지정하면 100,000 read를 가진 샘플도, 20,000 read를 가진 샘플에서 랜덤 하게 1,000 read을 추출하게 됩니다. 이후 다양성을 비교합니다.
그렇다면 이러한 랜덤추출방식은 왜 사용될까요?
바로 각 샘플의 depth가 다르기 때문입니다. 100,000 read를 가진 샘플은 당연하게도 1,000 read를 가진 샘플에 비해 다양성이 높게 나올 수 있습니다. 그러나 이러한 read생산은 실제 미생물의 풍부도를 반영하지 않고 여러 실험적인 과정에서 변동이 생길 수 있습니다. 그러니 이러한 변동을 줄이기 위해 약간 엄격하지만, 랜덤 하게 추출하여 비교하는 것입니다.
이러한 과정을 rarefy라고 합니다.
이때 데이터의 손실을 줄이기 위해 depth는 값이 클수록 좋겠죠?
그러나 depth를 너무 낮게 지정하면 퀄리티가 좋지 않은 서열이 분석에 사용되는 것이고, depth를 높게 하면 샘플의 수가 별로 남지 않으니 이 또한 퀄리티가 낮아질 수 있습니다.
depth를 정하는 건 까다로우니, 전 단계에서 만든 table.qzv 파일을 참고해야 합니다. 이왕이면 제외되는 서열이 적으면서 가능한 dpeth 높게 설정하는 것이 좋습니다(어렵죠?).
위 코드를 보면, 튜토리얼에서 최소 sampling depth는 1103으로 설정되었습니다. 왜 이러한 값으로 지정하였을까요?
table.qzv를 보시면 적어도 depth가 1,000 이상인 샘플의 depth가 1103이기 때문입니다.

즉, 이 연구자는 1,000 read이하를 가진 샘플은 버리더라고, 최대한 높은 depth를 선택하여 전체 샘플에서 추출하고자 하는 것입니다.
이때 아래 3개의 샘플은 제거됩니다.
실행 후 core-metrics-results 디렉터리에 저장된 파일들은 아래와 같습니다.

예시로 Beta diversity분석 중 bray_curtis_emperor.qzv 를 관찰해 보자.

오른쪽 사이드 창에서 여러 옵션을 변경하면 이런 식으로 body-site 간의 차이를 관찰할 수 있습니다.
07-1. alpha-group-significance
alpha diversity는 같은 group내의 다양성을 계산합니다.
위 튜토리얼에서 주요하게 봐야 하는 것은 같은 body-site에 속해있는 sample사이의 다양성입니다.
우리는 아래 명령어를 통해 faith_pd_vector.qza 파일을 분석하고 시각화해 봅시다.
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/faith_pd_vector.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization core-metrics-results/faith-pd-group-significance.qzv
faith-pd는 계통 간의 거리를 반영하는 다양성을 측정합니다.
faith-pd-group-significance.qzv
[ Alpha Diversity Boxplots ]
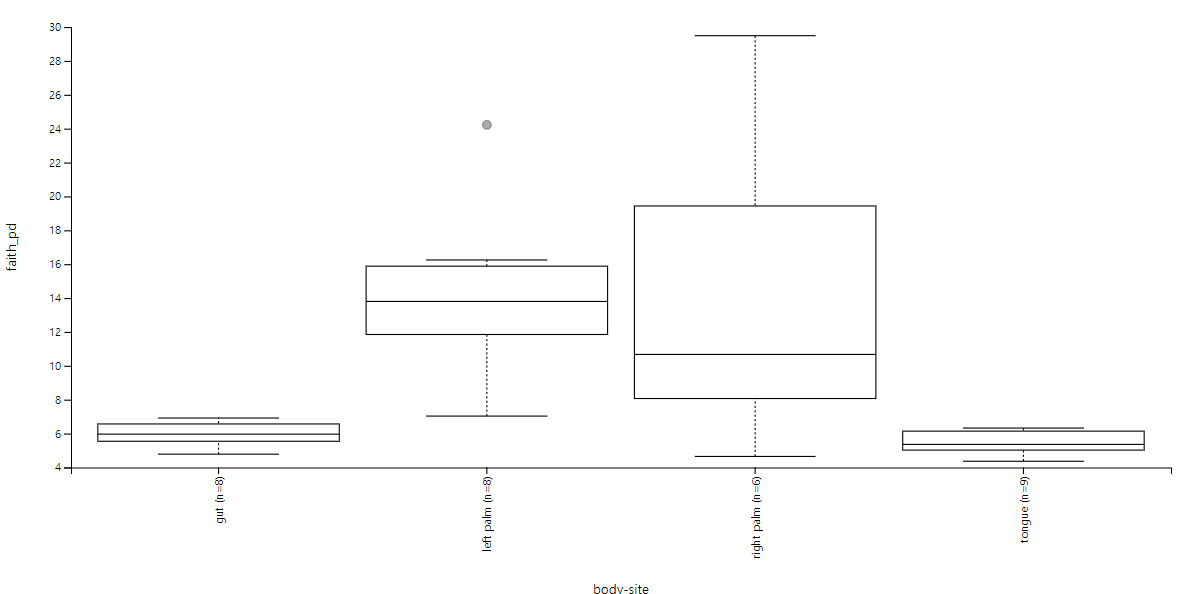
left palm, right palm의 다양성은 상대적으로 높고, gut과 tongue은 낮습니다.
[ Kruskal-Wallis (all groups) ]
| Result | |
| H | 18.82762096774192 |
| p-value | 0.00029677768053661975 |
3그룹 이상 차이를 보기 위해 Kruskal-Wallis분석이 수행되었습니다.
이때 p-value가 0.0002967는 0.05 이하임으로, 귀무가설을 기각하여 통계적으로 각 그룹마다 다양성이 다르다고 볼 수 있습니다.
[ Kruskal-Wallis (pairwise) ]
| Group1 | Group2 | H | p-value | q-value |
| gut (n=8) | left palm (n=8) | 11.29412 | 0.0007775 | 0.0023325 |
| gut (n=8) | right palm (n=6) | 4.266667 | 0.0388671 | 0.0583007 |
| gut (n=8) | tongue (n=9) | 3.342593 | 0.0675081 | 0.0810098 |
| left palm (n=8) | right palm (n=6) | 0.416667 | 0.5186050 | 0.5186050 |
| left palm (n=8) | tongue (n=9) | 12 | 0.0005320 | 0.0023326 |
| right palm (n=6) | tongue (n=9) | 5.555556 | 0.0184221 | 0.0368442 |
각 그룹 간의 비교값을 확인해 봅시다. 이때 우리는 q-value에 주목해야 합니다.
이를 보면 gut과 palm은 통계적으로 유의하게 차이가 나는 것을 알 수 있고, right-left palm 간의 유의한 차이가 나지 않는 것을 알 수 있습니다.
07-2. Beta-group-significance
beta diversity는 alpha와 다르게 그룹 간의 다양성을 봅니다.
계통 간의 관계를 고려하는 다양성지수는 unweighted-unifrac distance의 분석 결과를 확인해 봅시다.
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-column body-site \
--o-visualization core-metrics-results/unweighted-unifrac-body-site-significance.qzv \
--p-pairwise
--m-metadata-column : metadata안에 우리가 비교하길 원하는 그룹이 포함된 column 선택
--p-pairwise : 그룹 간 비교분석도 추가로 수행
--p-method : 분석방법은 'permanova', 'anosim', 'permdisp'에서 선택 가능하며 은 default로 'permanova'가 수행
결과 파일을 살펴보자.
unweighted-unifrac-body-site-significance.qzv

그룹 간 분석은 PERMANOVA분석이 수행되었으며 p-value를 보아 통계적으로 유의한 것을 볼 수 있습니다.
각 그룹 내에서 다양성을 확인해 봅시다.
Gut에서 각 부위의 샘플 간의 distance가 매우 큽니다. 이는 gut과 다른 부위의 마이크로바이옴은 상당히 이질적이라는 뜻입니다. 그러나 같은 gut 내에서는 비교적 샘플 간의 다양성(비유사성)이 크지 않습니다.


이후 다른 통계방법인 ANOSIM분석의 결과를 살펴봅시다.
qiime diversity beta-group-significance \
--i-distance-matrix weighted_unifrac_distance_matrix.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-column body-site \
--p-method anosim \
--output-dir weighted/beta-sig


위 분석에서는 --p-pairwise 옵션을 를 추가하지 않았으므로, 추가적인 pairwise 표는 제공되지 않습니다.
07-3. Principal Coordinates Analysis
이제 beta divercity의 시각화를 위한 PCoA plot을 그려보자.
이전에 core-metrics-phylogenetic 명령어로 (3D) Emperor plot 만들어서 보았습니다.
이제는 qiime emperor plot 명령어로 조금 더 세부적인 분석을 해볼까요?
# PCoA에서 날짜에 따른 축 추가
qiime emperor plot \
--i-pcoa core-metrics-results/unweighted_unifrac_pcoa_results.qza \
--m-metadata-file sample-metadata.tsv \
--p-custom-axes days-since-experiment-start \
--o-visualization core-metrics-results/unweighted-unifrac-emperor-days-since-experiment-start.qzv
# PCoA에서 날짜에 따른 축 추가
qiime emperor plot \
--i-pcoa core-metrics-results/bray_curtis_pcoa_results.qza \
--m-metadata-file sample-metadata.tsv \
--p-custom-axes days-since-experiment-start \
--o-visualization core-metrics-results/bray-curtis-emperor-days-since-experiment-start.qzv
각각의 결과 파일을 보자.
- unweighted-unifrac-emperor-days-since-experiment-start.qzv
- bray-curtis-emperor-days-since-experiment-start.qzv

오른쪽 사이드바에서 color에서 scatter : body-site를 적용하자. 날짜 별로 몸의 부위에 따른 plot이 그려집니다.
08. Alpha rarefaction plotting
rarefaction plot이란 각 샘플에서 read의 수(depth)가 증가됨에 따라 계산되는 Alpha diversity (즉 feature의 다양성)이 얼마나 증가하는지 표시한 그래프입니다.
이 그래프를 그리는 이유는, 적절한 depth가 되었을 때 샘플 내의 다양성(alpha)의 증가가 멈추는지 판별하기 위해서입니다.
만약 depth가 1,000에서 alpha 다양성 지수인 shannon지수의 기울기가 평행에 가까울 때, 이 샘플들은 1,000 depth이상만 가지고 있으면 그 샘플 내에서 볼 수 있는 모든 feature를 다 관찰했다고 볼 수 있습니다.
즉, 시퀀싱이 충분히 되었다!라고 볼 수 있는 거죠.
만약 1,000 depth에서 아직 shannon이 증가하고 있는 추세라면(우상향 선), 그 샘플은 depth가 더 증가할 때마다, 다양성이 더 증가하겠죠? 즉 더 많은 depth를 필요로 한다는 뜻입니다.
qiime diversity alpha-rarefaction \
--i-table table.qza \
--i-phylogeny rooted-tree.qza \
--p-max-depth 4000 \
--m-metadata-file sample-metadata.tsv \
--o-visualization alpha-rarefaction.qzv

각 body site로 관찰했을 때, 모든 부위의 시퀀싱은 충분히 진행되었음을 알 수 있습니다.

위 plot도 각 body Site에 따라서 시퀀싱 뎁스가 증가할 때, 각 depth에 해당하는 샘플의 수를 나타낸 것입니다
이렇게 각 샘플을 보는 것이 아니라 body Site처럼 그룹으로 샘플의 rarefaction을 확일 할 때, 추가적인 정보를 제공해 줍니다.
09. Taxonomic analysis
이제, 샘플마다 어떠한 계통을 가진 생물이 존재하는지 알아봅시다.
여기에 사용된 database는 greengene 13_8 ver. 을 사용됩니다. 이는 연구실에서 거의 사용되지 않는 오래된 데이터 베이스입니다.
2022년에 새로운 greengene이 출시되었지만, tutorial에서는 13년도 버전을 사용하네요ㅠ
데이터 베이스 다운로드하기
wget https://data.qiime2.org/2024.2/common/gg-13-8-99-515-806-nb-classifier.qza
- 각 featureID에 계통 정보 매칭시키기
qiime feature-classifier classify-sklearn \
--i-classifier gg-13-8-99-515-806-nb-classifier.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
결과 파일인 taxonomy.qzv을 관찰해 보자.

각 featureID에 매칭된 taxon정보와, 그 매칭의 신뢰도 점수를 보여주고 있습니다.
taxa bar로 시각화해 봅시다.
qiime taxa barplot \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization taxa-bar-plots.qzv
taxa-bar-plots.qzv에서, level을 level6(Genus-level)으로 바꾸어 관찰해 봅시다.
또한 샘플을 Body site와 항생제 사용유무에 따라 정렬해 봅시다.

장 샘플에서는 Bacteroides가 가장 풍부하며, 손바닥은 여러 Genus가 존재합니다.
혀에는 Streptococcus, Neisseria, Prevotella 등이 풍부하게 존재하는 것을 볼 수 있습니다.
10. Differential abundance testing with ANCOM
그렇다면 각 그룹을 대표하는 특이 마이크로바이옴이 무엇인지 알아보기 위한 방법은 무엇일까요?
이는 차등발현분석(Differential abundance analysis;DAA)으로 수행됩니다.
DAA에는 여러 방법(LEfse, ANCOM, edgeR..)이 있으나 그중에서 ANCOM2를 사용합니다. ANCOM 방법에 대한 장단점이 있으며 이는 논문(ANCOM paper)에서 확인할 수 있습니다. ANCOM은 25% 이하의 feature가 그룹 간 차이가 날 것이라고 가정하여 분석하는 방법입니다. (ANCOM2 분석을 위한 qiime plugin에는 두 종류가 있는데 우리는 q2-composition을 사용합니다.)
이 튜토리얼에서는 두 사람의 데이터가 포함되어 있습니다. 이때 장 마이크로바이옴에서 두 사람 사이에 차이가 있는 미생물을 비교해 봅시다.
qiime feature-table filter-samples \
--i-table table.qza \
--m-metadata-file sample-metadata.tsv \
--p-where "[body-site]='gut'" \
--o-filtered-table gut-table.qza
gut-table.qza은 relative abundance가 아닌 count table(FeatureTable [Frequency] ) 데이터를 가집니다.
ancom분석을 위해 FeatureTable [Composition]로 바꾸어 줍시다.
qiime composition add-pseudocount \
--i-table gut-table.qza \
--o-composition-table comp-gut-table.qza
| count per sample table vs relative abundance table
count per sample table
| Sample_1 | Sample_2 | Sample_3 | |
| OTU_1 | 15 | 50 | 10 |
| OTU_2 | 15 | 20 | 4 |
| OTU_3 | 20 | 30 | 6 |
relative abundance table
- 샘플에 read 합(depth)으로 각 OTU에 해당하는 read값을 나누어 준다. 각 값은 0에서 1 사이 값을 가집니다,
- 미생물 데이터는 샘플마다 depth가 다르기 때문에, 이를 표준화해 주기 위해 실행합니다.
- 또한 NGS데이터는 샘플 종류, 시퀀싱 플랫폼, 시퀀싱 run마다 결과물이 다르다. 이를 보정해 주는 역할도 합니다.
| Sample_1 | Sample_2 | Sample_3 | |
| OTU_1 | 0.3 | 0.5 | 0.5 |
| OTU_2 | 0.3 | 0.2 | 0.2 |
| OTU_3 | 0.4 | 0.3 | 0.3 |
이제 ANCOM 분석 결과를 관찰해 봅시다.
qiime composition ancom \
--i-table comp-gut-table.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-column subject \
--o-visualization ancom-subject.qzv
ancom-subject.qzv을관찰해 보자. 아래와 같은 그림을 Volcano plot이라고 부릅니다. Volcano plot 이란 V자모양으로 샘플들이 퍼져있습니다.
여기서 w값이 을 기준으로 차등적인 발현도가 높을수록 가장자리의 윗부분에 위치합니다.

ANCOM 분석에서는 relative abundance로 변환된 OTU table이 Central log ration(clr)로 변환된다.
ANCOM의 귀무가설은 "한 그룹 내 특정 종의 평균 풍부도가 다른 그룹과 동일하다"이며, w값은 귀무가설이 기각된 횟수를 나타냅니다. ANCOM 분석은 p-value값을 반환하지 않습니다. 대신 default로 w값이 가장 큰 샘플에서 상위 30% 이하면 유의하다고 판단합니다.
각 feature 각 위치하는 곳이 음수인 x 값이면, Subject-1에서 발현이 많다는 것이고, 양수의 x값은 Subject-2에서 발현이 많다는 것을 뜻합니다.

하지만 저 featureID가 어떤 taxa 인지 알아야겠죠? 관련 데이터를 확인해 봅시다.
qiime taxa collapse \ # taxonomy 부착
--i-table gut-table.qza \
--i-taxonomy taxonomy.qza \
--p-level 6 \
--o-collapsed-table gut-table-L6.qza
# ANCOM 분석 수행
qiime composition add-pseudocount \
--i-table gut-table-L6.qza \
--o-composition-table comp-gut-table-L6.qza
qiime composition ancom \
--i-table comp-gut-table-L6.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-column subject \
--o-visualization L6-ancom-subject.qzv
결과 파일인 L6-ancom-subject.qzv를 확인해 보자 (L6= Genus-level).
이는 각 feature level(ASV level)로 본 것이 아니라, Genus level로 계통을 합쳐서 본 결과입니다.


결과적으로 Parabacteroides가 subject-2에서 차등적으로 존재하는 것을 확인할 수 있었습니다.
자 이렇게 마이크로바이옴 기본 분석 단계인 alpha, beta diversity와 taxonomy composition, DAA를 수행해 보았습니다.
대부분의 마이크로바이옴 분석은 R 상에서 이루어집니다,
그렇다면, QIIME2의 데이터를 R로 옮겨서 자세한 시각화를 수행해 볼까요?
[qiime2r] QIIME2의 qza파일을 R의 phyloseq으로 간편하게 변환하기
| qiime2R 설치 if (!requireNamespace("devtools", quietly = TRUE)){install.packages("devtools")} devtools::install_github("jbisanz/qiime2R") # current version is 0.99.20 | 예제 데이터 - 출처 : https://docs.qiime2.org/2020.2/tutorials/moving-picture
bio-kcs.tistory.com
[Phyloseq] phyloseq을 이용해 phylum별로, sample type별로 NMDS plot그리기
[Microbiome분석 단계] 1) Preprocessing decontam 2) OTU clustering 3) Taxonomy classification 1 4) Diversity - Alpha diversity - Beta diversity 1 6) Differential abundance analysis 7) Functional analysis 8) Network analysis(Correlation) 1 +) Machine lea
bio-kcs.tistory.com
[Phyloseq] phyloseq data로 Beta diversity에서 PERMANOVA 분석하기
👩💻 Microbiome분석 단계 1) Preprocessing decontam 2) OTU clustering 3) Taxonomy classification 1 2 4) Diversity - Alpha diversity - Beta diversity 1 6) Differential abundance analysis 7) Functional analysis 8) Network analysis(Correlation) 1 +)
bio-kcs.tistory.com
[Phyloseq] R에서 phyloseq 데이터로 LefSe 분석하기(micobiomeMarker/microbial 패키지 사용) + 에러
작성 : 2022-10-24 수정 : 2023-06-04 (microbial 패키지 추가) 🟦 목표 1. Microbiome의 marker 미생물을 찾는데 많이 사용되는 LefSe 분석에 대해 알아보고 2. R을 이용하여 분석을 후 시각화해 보자 🟦 LefSe 분석
bio-kcs.tistory.com
[R/Phyloseq] Taxonomy composition plot에서 Phylum별로 Genus에 색 변화 주는 3가지 방법
- 작성 시작 : 2023-01-17 - 작성 완료 : 2023-02-28 논문에서는 독창성이 가장 가치가 높은 가치로 꼽히지만, 그러한 내용을 설명하는 그림자료도에도 신경을 써야 한다. 마이크로바이옴 분야에서 흔하
bio-kcs.tistory.com
[R/phyloseq] Phyloseq Filtering 방법 10가지 정리
🟦 샘플 필터링은 왜 필요한가? ◼ 마이크로 바이옴 분석에서 데이터는 보통 대용량 데이터이다. ◼ 분석 시 컴퓨터의 과부하를 줄이기 위하여 일부 데이터만 추출하여 비교하기도 한다. - ex) ma
bio-kcs.tistory.com
[R/Phyloseq] 각 Phylum이 몇 퍼센트를 차지하는지 보여주는 함수
각 Phylum이 몇 퍼센트를 차지하는지 계산해 보자. 데이터는 Phyloseq의 기본 데이터 GlobalPatterns을 사용한다. | 데이터 불러오기 library(phyloseq) library(dplyr) data("GlobalPatterns") GlobalPatterns # read count table ->
bio-kcs.tistory.com
[R] Microbiome데이터를 사용한 Regression plot 그리기 +) phyloseq
작성 시작 : 2023-05-16 ~ 2023-05-17 Regression 은 주로 연속형 데이터의 예측에 사용된다. 아래는 Microbiome 분야에서 사용되는 regression plot의 종류를 알아보자 사실 원하는 두 데이터만 있으면 linear regress
bio-kcs.tistory.com
[R] Microbiome데이터에서 correlation plot 그리기 +) Phyloseq 개체 사용하기
작성 : 2023.06.21 | 목표 마이크로바이옴 데이터로 여러 종류의 상관관계 그림을 그려보자. 특히 Heatmap에 집중해서 관찰하자! | 예제 데이터 - qiime2 moving pictures Tutorial에 나오는 데이터로, 사람의 4
bio-kcs.tistory.com
[R] Core microbiome 분석 하기
| Core microbiome 이란? 샘플에서 가장 주요한 마이크로바이옴을 분석하는 방법으로 주로, 아래와 같은 그림으로 나타낸다. Core microbiome의 개념은 아직 정립되지 않았지만 주로 전체 샘플에서 많은
bio-kcs.tistory.com
[Metacoder] Heat tree그리는 방법
작성: 2023-08-25 Metacoder란? - 공식 튜토리얼 : https://grunwaldlab.github.io/metacoder_documentation/workshop--05--plotting.html - 논문 : Foster, Z. S., Sharpton, T. J., & Grünwald, N. J. (2017). Metacoder: An R package for visualization and m
bio-kcs.tistory.com
[microbiomeMarker] 마이크로바이옴 데이터에 머신러닝을 적용해보자(매우쉬움)
- 작성 : 2023.08.29 1. MicrobiomeMarker 패키지에 대해서 - 논문 : Cao, Y., Dong, Q., Wang, D., Zhang, P., Liu, Y., & Niu, C. (2022). microbiomeMarker: an R/Bioconductor package for microbiome marker identification and visualization. Bioinformatic
bio-kcs.tistory.com
[Vegan] phyloseq 개체를 이용해서 biplot그리기
작성 2023.09.04 수정 2023.09.26 Vegan - Vegan 패키지는 환경데이터를 처리 및 분석에 사용된다. 마이크로바이옴 데이터와 환경데이터는 샘플이름이 열에, 환경 또는 미생물의 이름이 행에 위치(혹은 그
bio-kcs.tistory.com
[Maaslin2] phyloseq을 사용해 Maaslin2에서 3그룹 간 비교하기
Maaslin2(Microbiome Multivariable Associations with Linear Models)이란? - 홈페이지 : https://huttenhower.sph.harvard.edu/maaslin/ - Lefse를 개발한 하버드 연구소에서 만든 Differential abundance analysis 도구이다. R package로 제공
bio-kcs.tistory.com
[R] alpha diveristy의 각 index의 Mean, Sd, N, Upper, Lower, 95% CI값과 통계 분석 결과의 p-value값 저장하기
필요 데이터 - phyloseq 목표 결과 - 각 alpha diversity 간의 통계 분석 자료 제작 이유 - 엑셀로 정리하다가 코딩으로 만들면 훨씬 간편할 것 같아서 - 다 만들고 나니 정말로 그랬다. #### 필요 library(phyl
bio-kcs.tistory.com
[R] Abundance의 평균, 표준편차, N, 95% CI(upper, lower)값 계산해주는 함수
작성: 2023.10.13 논문을 쓸 때, 특정 taxa에 대한 구체적인 abundance값을 기입하는 경우가 많습니다. 보통 "mean: 38.9%, SD 4.3 %"의 형태로 많이 작성되는데요, 이 값들은 어떻게 구하는 것이 좋을까요? 1.
bio-kcs.tistory.com
[R/phyloseq] Neutral community model(NCM) 그리고 해석하기
작성: 2023-10-16-월 | Sloan's neutral community model(NCM) 이란? NCM은 환경 생태쪽에서 발전한 개념으로 모든 종이 환경에서 중립적이라는 전제를 가지고 있다. 이 모델의 주요 특징은 아래와 같다. 1. 중립
bio-kcs.tistory.com
[NetCoMi] phyloseq으로 spearman correlation과 sparCC로 network plot을 그려보자.
글 작성: 24.01.03. 내용추가: 24.01.02. 1. Network analysis | 마이크로바이옴 연구에서 네트워크 연구란? - microbiome은 복잡한 미생물 군집을 말한다. 여러 생물들의 상호작용은 전체 미생물의 구조를 안정
bio-kcs.tistory.com
[R] PCoA 에 가장자리 plot 추가하는 5가지 방법 The five methods for beta diversity side panel
작성: 2024-10-21 수정: 2024-01-08 side panel 은 beta diversity를 나타내는 PCA, PCoA 그림의 추가 정보를 나타냅니다. PCoA의 side panel 은 각 샘플의 dissimilarity 간의 분포를 축소해서 나타내는 PCoA 의 축 1과 2를 b
bio-kcs.tistory.com
[R] Venn diagram을 그리는 패키지 5개 비교
Venn diagram은 각 그룹에 어떠한 공통적인 속성이 있는지를 한눈에 보여줍니다. R에서는 여러 가지 도구에 의해서 제공됩니다. 그렇다 보니 어떤 도구를 선택해야 할지 많은 고민이 있습니다. 제가
bio-kcs.tistory.com
Reference
- [KGOL] 마이크로바이옴 분석 (경희대 이재형)
- https://docs.qiime2.org/2022.2/tutorials/moving-pictures/#demultiplexing-sequences