
- 작성 시작 : 2023-01-17
- 작성 완료 : 2023-02-28
논문에서는 독창성이 가장 가치가 높은 가치로 꼽히지만, 그러한 내용을 설명하는 그림자료도에도 신경을 써야 한다.
마이크로바이옴 분야에서 흔하게 사용되는 figure는 alpha diversity, beta divertsity, Taxonomy composition이 있다.
그 중 Taxonomy composition은 각 샘플에서 어떤 taxa가 상대적인 분포를 갖는지 보여주는 그림이다.
이 글에서는 phyloseq 예제 데이터인 GlobalPatterns을 사용해서 시각화 방법 3가지를 소개한다.
🟦 1. 수동으로 RColorBrewer색 지정
- Phyloseq 객채 : GlobalPatterns
- 내가 기존에 사용하던 방법으로, 가장 손이 많이 가는 방법이다.
◼ 데이터 준비하기
library(reshape2)
library(phyloseq)
library(ggplot2)
library(dplyr)
library(RColorBrewer)
data("GlobalPatterns")
Global.melt <- GlobalPatterns %>% tax_glom(taxrank = 'Family') %>% psmelt()
◼ Top 10 Family 보기
Order = tapply(Global.melt2$Abundance, Global.melt2$Family, sum) %>% sort(decreasing = T) # abundance 순으로 정렬
Top10 <- names(Order[c(1:10)])
# [1] "ACK-M1" "Bacteroidaceae" "Enterobacteriaceae" "Flavobacteriaceae" "Lachnospiraceae" "Neisseriaceae" "Nostocaceae"
# [8] "Pasteurellaceae" "Ruminococcaceae" "Streptococcaceae"
Global.melt2 <- Global.melt2[order(-Global.melt2$Abundance),]
a <- Global.melt2[Global.melt2$Family %in% Top10, c("Abundance", "Phylum", "Family")]
a.1 <- a %>% select(Phylum, Family) %>% .[!duplicated(.$Family),]
# Bacteroidetes Bacteroidaceae
# Cyanobacteria Nostocaceae
# Proteobacteria Neisseriaceae
# Firmicutes Ruminococcaceae
# Actinobacteria ACK-M1
# Bacteroidetes Flavobacteriaceae
# Proteobacteria Pasteurellaceae
# Firmicutes Lachnospiraceae
# Proteobacteria Enterobacteriaceae
# Firmicutes Streptococcaceae
# 수동으로 순서 지정하기
Top10_SortByph <- c(# Actinobacteria
"ACK-M1",
"Other Actinobacteria",
# Bacteroidetes
"Bacteroidaceae", "Flavobacteriaceae",
"Other Bacteroidetes",
# Cyanobacteria
"Nostocaceae",
"Other Cyanobacteria",
# Firmicutes
"Ruminococcaceae", "Lachnospiraceae", "Streptococcaceae",
"Other Firmicutes",
# Proteobacteria
"Neisseriaceae", "Pasteurellaceae", "Enterobacteriaceae",
"Other Proteobacteria",
"Others")
◼ Others 처리
- 각 Phylum에서 top10을 제외한 family를 Other로 바꾸어 주기
Global.melt2$Family[Global.melt2$Phylum=="Actinobacteria" &
!Global.melt2$Family %in% c('ACK-M1')] <- "Other Actinobacteria"
Global.melt2$Family[Global.melt2$Phylum=="Bacteroidetes" &
!Global.melt2$Family %in% c( "Bacteroidaceae", "Flavobacteriaceae")] <- "Other Bacteroidetes"
Global.melt2$Family[Global.melt2$Phylum=="Cyanobacteria" &
!Global.melt2$Family %in% c( "Nostocaceae")] <- "Other Cyanobacteria"
Global.melt2$Family[Global.melt2$Phylum=="Firmicutes" &
!Global.melt2$Family %in% c("Ruminococcaceae", "Lachnospiraceae", "Streptococcaceae")] <- "Other Firmicutes"
Global.melt2$Family[Global.melt2$Phylum=="Proteobacteria" &
!Global.melt2$Family %in% c("Neisseriaceae", "Pasteurellaceae", "Enterobacteriaceae")] <- "Other Proteobacteria"
# 아까 지정한 family 순서 적용하기 (factor 이용)
Global.melt2$Family <- factor(Global.melt2$Family, levels = Top10_SortByph)
unique(Global.melt2$Family) # 16 level
# [1] Bacteroidaceae Nostocaceae Neisseriaceae Ruminococcaceae ACK-M1
# [6] Flavobacteriaceae Pasteurellaceae Lachnospiraceae <NA> Enterobacteriaceae
# [11] Other Proteobacteria Streptococcaceae Other Bacteroidetes Other Actinobacteria Other Firmicutes
# [16] Other Cyanobacteria
# 16 Levels: ACK-M1 Other Actinobacteria Bacteroidaceae Flavobacteriaceae Other Bacteroidetes ... Others
◼ 수동으로 색 지정하기
col <- c("#E78AC3","#E74AC3",
brewer.pal(3, "Blues"),
"#FDAE6B", "#E6550D",
brewer.pal(4, "Greens"),
brewer.pal(4, "BuPu"),
"#BDBDBD")- 각 Phylum별로 색을 다르게 지정하였으며, RColorBrewer 패키지를 사용하여 색이 잘 구분되게 그렸다.
◼ bar plot
p <- ggplot(data=Global.melt2, aes(x=Sample, y=Abundance, fill=Family)) +
geom_bar(aes(), position="fill", stat="identity") +
scale_fill_manual(values = col) +
theme_classic() +
theme(legend.position="bottom" ,
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_blank()) +
labs(y = "Relative Abundance (%)") +
theme(axis.text.x.bottom = element_text(angle = 45, vjust = 1, hjust=1),
axis.text = element_text(size = 7))
p
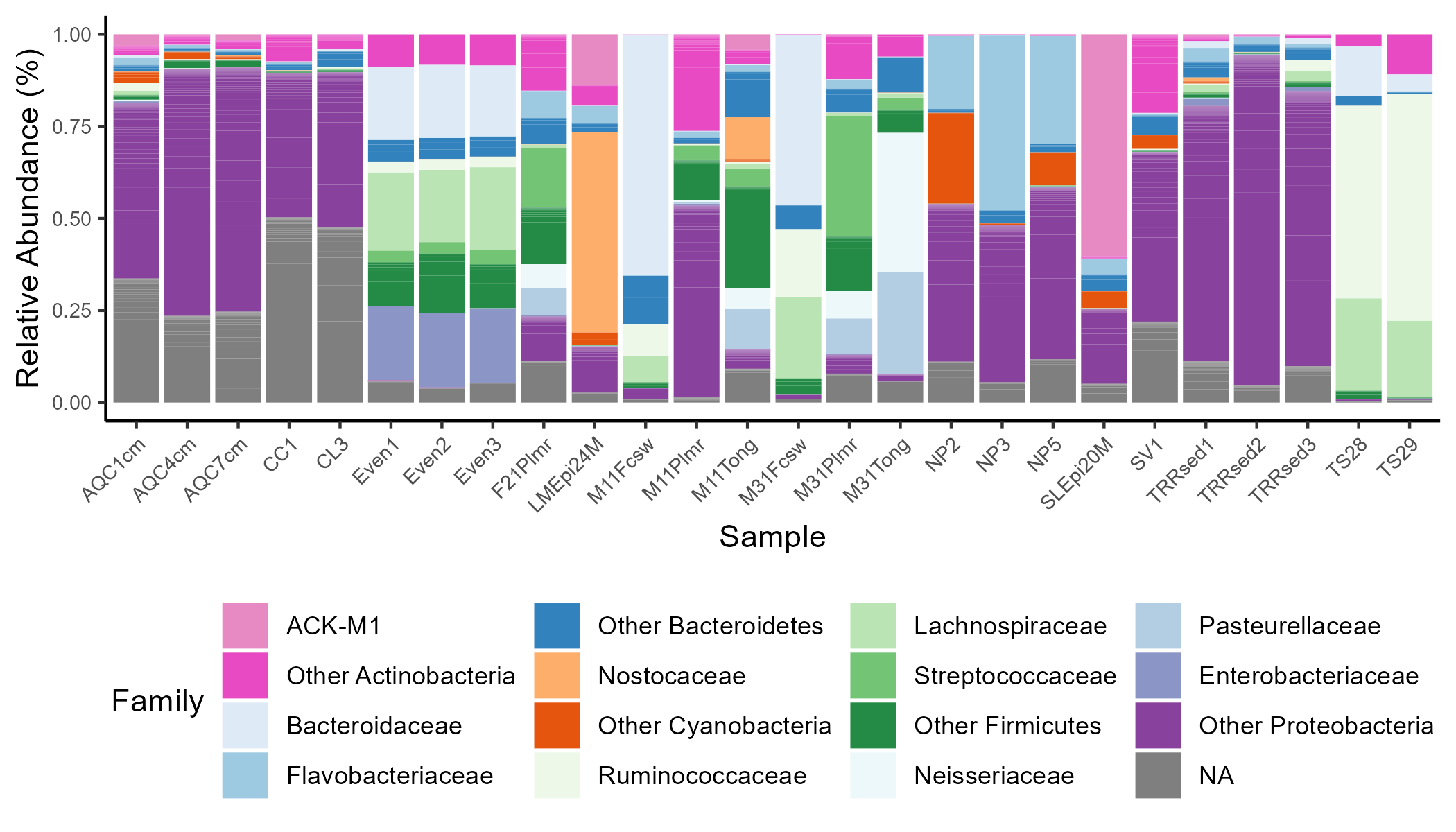
- 실수로 NA값을 처리하지 못했지만, family와 phylum level을 모두 나타내는 그림이다.
- 그러나 Phylum level을 모두 나타내지는 못했다 이는 Others에 포함되기 때문이다.
◼ 장점
- 가장 뚜렷한 색 구분으로 보는 이에게 확실한 의미를 전달할 수 있다
◼ 단점
- 손으로 수정해야 하는 부분이 가장 많음으로 번거로울 수 있다. (매우 귀찮다)
🟦 2. Phylum에 따라 색 나누는 hue_pal 함수 이용
◼ 패키지와 데이터 불러오기
library(phyloseq)
library(ggplot2)
library(dplyr)
library(RColorBrewer)
data("GlobalPatterns")
GlobalPatterns
df <- psmelt(GlobalPatterns)◼ Others 처리하기
df1 <- df
phylums <- c('Proteobacteria','Bacteroidetes','Firmicutes')
df1$Phylum[!df1$Phylum %in% phylums] <- "Others"
df1$Family[!df1$Phylum %in% phylums] <- "Others"
df1$Family[df1$Phylum=="Proteobacteria" &
!df1$Family %in% c('Alcaligenaceae','Enterobacteriaceae')] <- "Other Protobacteria"
df1$Family[df1$Phylum=="Bacteroidetes" &
!df1$Family %in% c('Bacteroidaceae','Rikenellaceae','Porphyromonadaceae')] <- "Other Bacteroidetes"
df1$Family[df1$Phylum=="Firmicutes" &
!df1$Family %in% c('Lactobacillaceae','Clostridiaceae','Ruminococcaceae','Lachnospiraceae')] <- "Other Firmicutes"
◼ 색을 지정해 줄 Phylum과 Family 추출하기
library(forcats)
library(dplyr)
df2 <- select(df1, Sample, Phylum, Family) %>%
mutate(Phylum=factor(Phylum, levels=c(phylums, "Others")),
Family=fct_reorder(Family, 10*as.integer(Phylum) + grepl("Others", Family))) %>%
group_by(Family) %>% # For this dataset only
sample_n(100) # Otherwise, unnecessary
df2
# Sample Phylum Family
# TRRsed3 Proteobacteria Alcaligenaceae
# TRRsed3 Proteobacteria Alcaligenaceae
# TRRsed3 Proteobacteria Alcaligenaceae
# TRRsed1 Proteobacteria Alcaligenaceae
# F21Plmr Proteobacteria Alcaligenaceae
# SV1 Proteobacteria Alcaligenaceae
# NP2 Proteobacteria Alcaligenaceae
# Even2 Proteobacteria Alcaligenaceae
# CL3 Proteobacteria Alcaligenaceae
# M31Fcsw Proteobacteria Alcaligenaceae
◼ 색 할당하기
# 출처 : https://stackoverflow.com/questions/49818271/stacked-barplot-with-colour-gradients-for-each-bar?rq=1
ColourPalleteMulti <- function(df, group, subgroup){
# Find how many colour categories to create and the number of colours in each
categories <- aggregate(as.formula(paste(subgroup, group, sep="~" )), df, function(x) length(unique(x)))
category.start <- (scales::hue_pal(l = 100)(nrow(categories))) # Set the top of the colour pallete
category.end <- (scales::hue_pal(l = 40)(nrow(categories))) # set the bottom
# Build Colour pallette
colours <- unlist(lapply(1:nrow(categories),
function(i){
colorRampPalette(colors = c(category.start[i], category.end[i]))(categories[i,2])}))
return(colours)
}
colours <- ColourPalleteMulti(df2, "Phylum", "Family")
◼시각화하기
library(ggplot2)
ggplot(df2, aes(x=Sample, fill = Family)) +
geom_bar(position="fill", colour = "grey") + # Stacked 100% barplot
scale_fill_manual("", values=colours) +
theme(axis.text.x=element_text(angle=90, vjust=0.5)) + # Vertical x-axis tick labels
scale_y_continuous(labels = scales::percent_format()) +
labs(y="Relative abundance")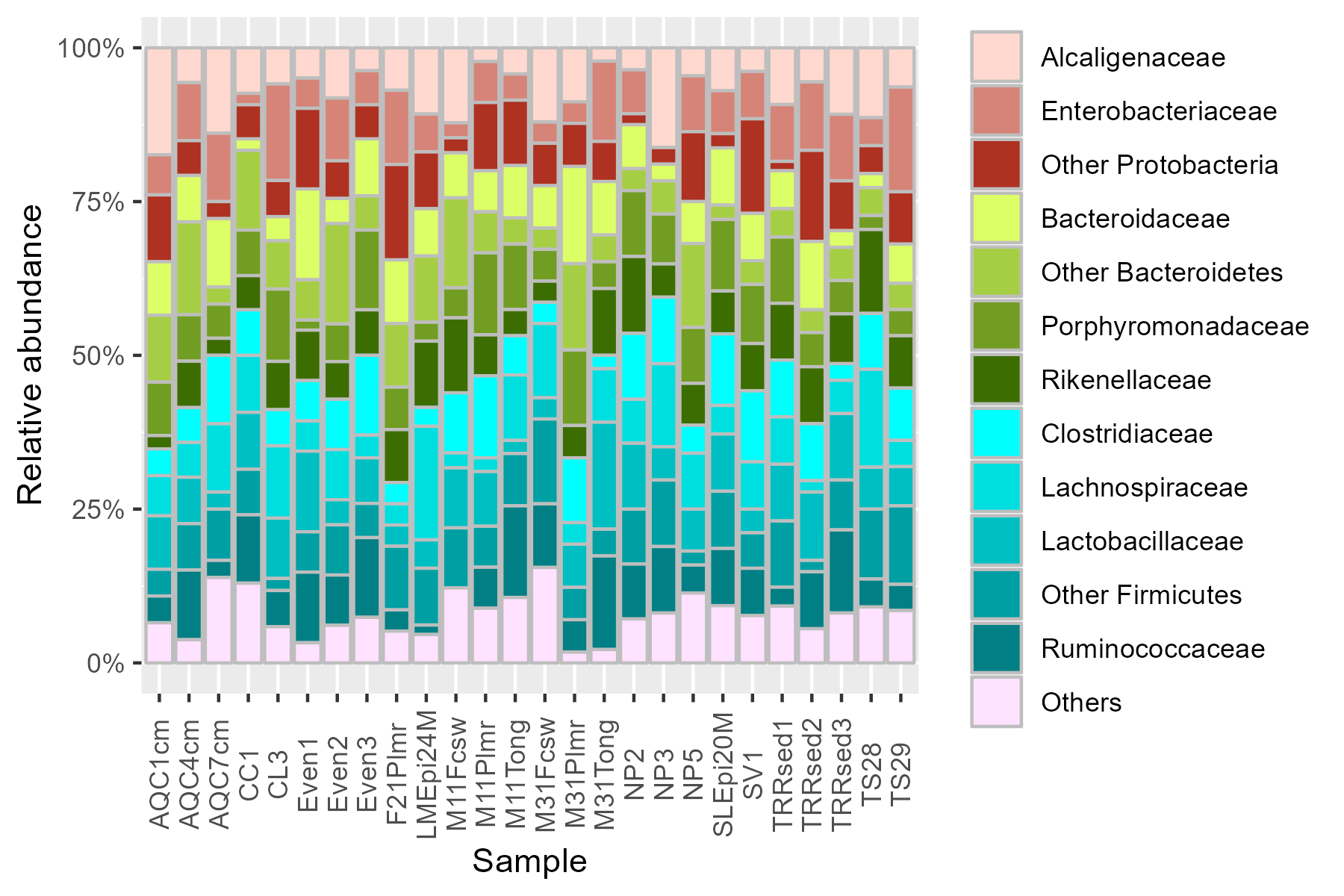
◼ 장점
- 각 Phylum 단계의 Others 도 포함하고 있어서 완전한 Phylum과 family의 정보까지 보여주는 그림이라고 할 수 있다.
◼ 단점
- Phylum에서 Others에 해당하는 것을 일일이 지정해 주어야 한다.
- 같은 Phylum에 여러 family가 있으면 색의 구분이 어려울 것이다.
🟦 3. fantaxtic package
- 🏠 : https://github.com/gmteunisse/Fantaxtic
- Phyloseq 객채에서 Taxonomy plot 그림에 전문화된 package로 손쉽게 원하는 taxa에 색 gradient를 줄 수 있다.
◼ Install
devtools::install_github("gmteunisse/fantaxtic")
require("fantaxtic")
require("phyloseq")
library(tidyverse)◼ 색을 지정할 Taxa level 결정
top_nested <- nested_top_taxa(GlobalPatterns,
top_tax_level = "Phylum",
nested_tax_level = "Genus",
n_top_taxa = 3,
n_nested_taxa = 3)- top_tax_level = "Phylum" # 메인 색을 정할 top taxa level
- nested_tax_level = "Genus" # 색의 변화로 표현할 nested taxa level
n_top_taxa = 3, # top taxa level의 수
n_nested_taxa = 3 # nested taxa level의 수
◼ Phylum의 total abundance순으로 정렬
- 위 예시에서는 Proteobacteria를 기준을 정렬하겠다.
# Order samples by the total abundance of Proteobacteria
sample_order <- psmelt(top_nested$ps_obj) %>%
data.frame() %>%
# relative abundances 계산
group_by(Sample) %>%
mutate(Abundance = Abundance / sum(Abundance)) %>%
# 원하는 Phylum level로 정렬한다
filter(Phylum == "Proteobacteria") %>%
group_by(Sample) %>%
summarise(Abundance = sum(Abundance)) %>%
arrange(Abundance) %>%
# Extract the sample order
pull(Sample) %>% as.character()
sample_order
# [1] "TS29" "TS28" "M31Fcsw" "M11Fcsw" "LMEpi24M" "AQC1cm" "AQC4cm" "SLEpi20M"
# [9] "Even2" "Even3" "Even1" "M11Tong" "M31Plmr" "AQC7cm" "F21Plmr" "NP5"
# [17] "CC1" "SV1" "CL3" "NP3" "NP2" "M11Plmr" "TRRsed1" "M31Tong"
# [25] "TRRsed3" "TRRsed2"
◼ 시각화
# Plot
p <- plot_nested_bar(top_nested$ps_obj,
"Phylum",
"Genus",
sample_order = sample_order,
nested_merged_label = "NA and other")
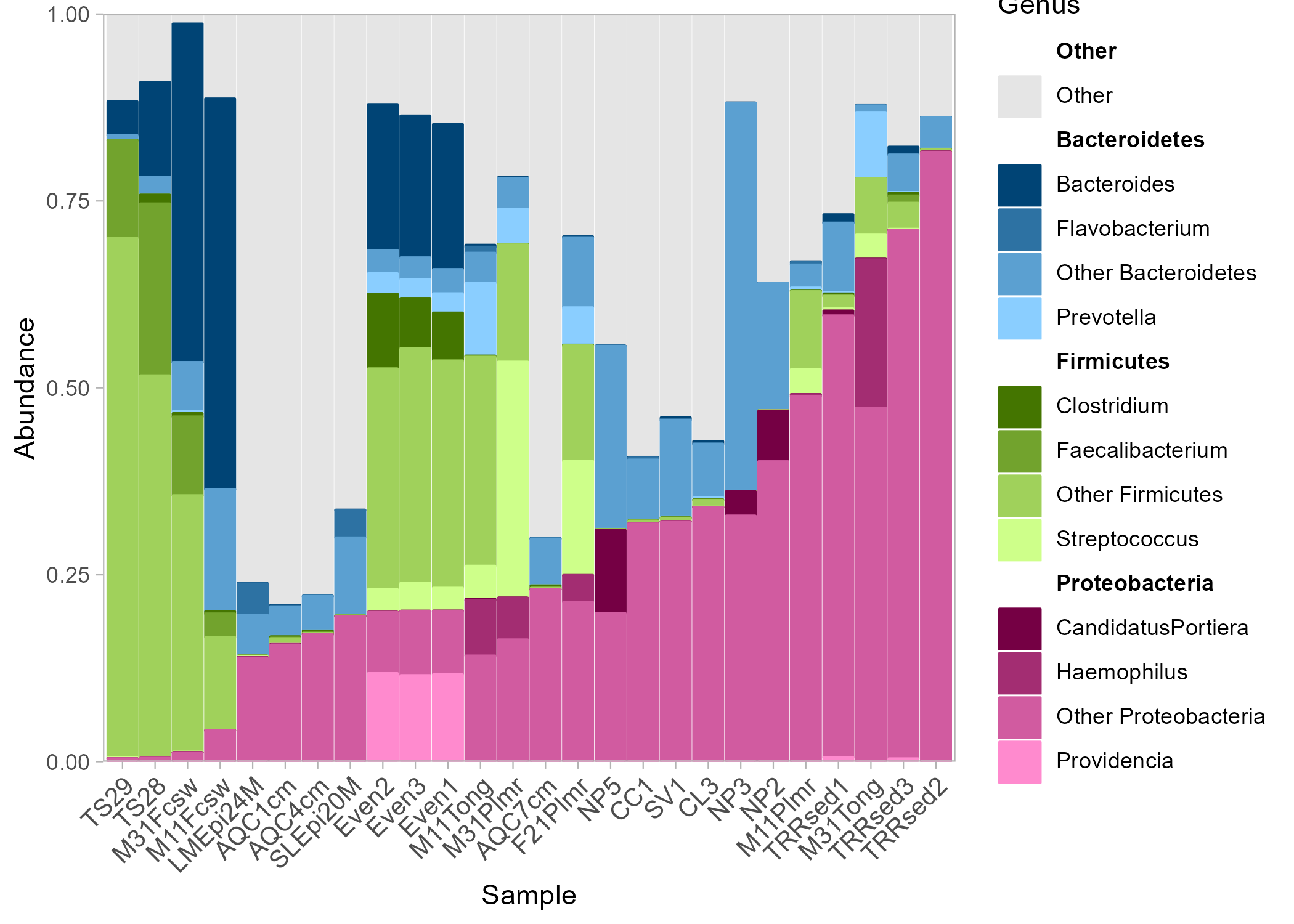
p + labs(title = "Relative abundances of the top 3 species for each of the top 3 phyla")
p + theme(panel.background = element_rect(fill='transparent'),
plot.background = element_rect(fill='transparent', color=NA),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.text.x = element_text(hjust = 1, vjust = 1, angle = 45, size = 10)
)
p + facet_wrap(~SampleType, scales = "free_x")이렇게 facet_wrap()을 이용해 SampleType별로도 관찰이 가능하다
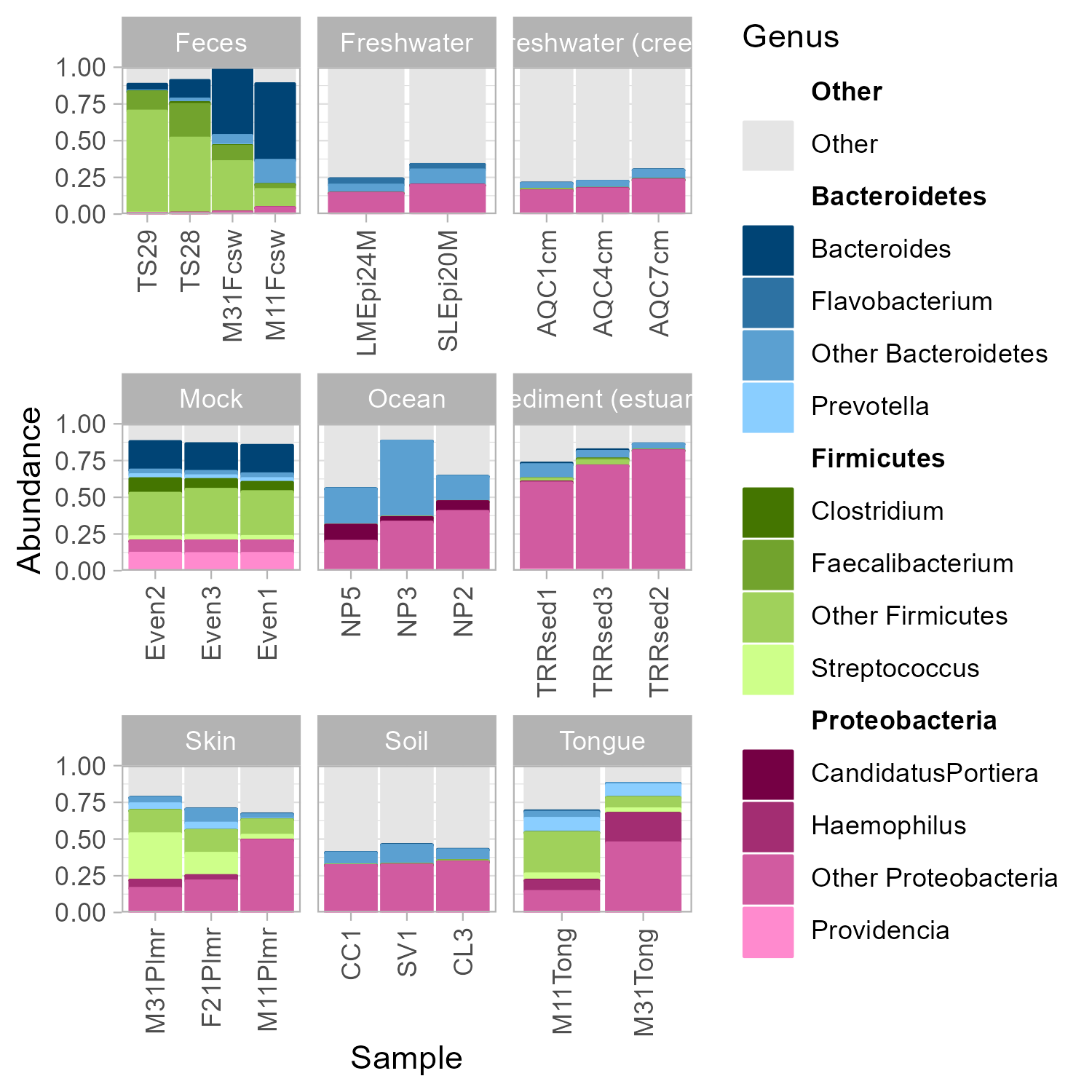
◼ 장점
- 가장 간단한 코드로 깔끔한 그림을 얻을 수 있다.
- Top taxa를 여러 taxa 레벨로 나누어서 색을 자동 지정해 주기 때문에 시각화 시 매우 편리하다.
◼ 단점
- 실제로 사용해 본 결과, 각 Phylum에 색이 5가지 이상이면 ggnested함수가 이를 인식하고, 위 패키지의 기본 색이 부여되는데, 이 색도 2번 방법처럼 각 taxa level마다 정확한 색 구분이 어렵다.
- 또한 ggnested 함수로 넘어가면 Phylum와 Genus 레벨을 보여주는 것이 아닌, Genus level만 보여주고 있지만, index의 카테고리는 Phylum level이 적혀있다. 이는 보는 이로 하여금 위 그래프가 Phylum 레벨의 내용을 모두 설명하는 것처럼 보일 수도 있다.
- 또한 index 간의 위치를 바꾸는 것이 어렵고, Phylum 레벨에서만 바꾸는 것이 가능하다.
🟦 결론
◼ 편한 방법만이 좋은 것은 아니다.
- 3번째 패키지가 가장 사용하기 편리하지만, 생각보다 제한이 많아서 자주 사용하게 되지는 않는 것 같다.
- 수동으로 일일이 지정하는 것은 귀찮지만 함수화 하여 필요할 때마다 불러오는 것이 훨씬 효과적일 것이다. 초기에 시간을 들여 사용한 함수들은 나중에 시간활용 부분에서 큰 도움이 된다. 그러므로 다시 만들러 가자..ㅠ
🟦 Reference
- https://github.com/gmteunisse/Fantaxtic
- 작성 시작 : 2023-01-17
- 작성 완료 : 2023-02-28
논문에서는 독창성이 가장 가치가 높은 가치로 꼽히지만, 그러한 내용을 설명하는 그림자료도에도 신경을 써야 한다.
마이크로바이옴 분야에서 흔하게 사용되는 figure는 alpha diversity, beta divertsity, Taxonomy composition이 있다.
그 중 Taxonomy composition은 각 샘플에서 어떤 taxa가 상대적인 분포를 갖는지 보여주는 그림이다.
이 글에서는 phyloseq 예제 데이터인 GlobalPatterns을 사용해서 시각화 방법 3가지를 소개한다.
🟦 1. 수동으로 RColorBrewer색 지정
- Phyloseq 객채 : GlobalPatterns
- 내가 기존에 사용하던 방법으로, 가장 손이 많이 가는 방법이다.
◼ 데이터 준비하기
library(reshape2)
library(phyloseq)
library(ggplot2)
library(dplyr)
library(RColorBrewer)
data("GlobalPatterns")
Global.melt <- GlobalPatterns %>% tax_glom(taxrank = 'Family') %>% psmelt()
◼ Top 10 Family 보기
Order = tapply(Global.melt2$Abundance, Global.melt2$Family, sum) %>% sort(decreasing = T) # abundance 순으로 정렬
Top10 <- names(Order[c(1:10)])
# [1] "ACK-M1" "Bacteroidaceae" "Enterobacteriaceae" "Flavobacteriaceae" "Lachnospiraceae" "Neisseriaceae" "Nostocaceae"
# [8] "Pasteurellaceae" "Ruminococcaceae" "Streptococcaceae"
Global.melt2 <- Global.melt2[order(-Global.melt2$Abundance),]
a <- Global.melt2[Global.melt2$Family %in% Top10, c("Abundance", "Phylum", "Family")]
a.1 <- a %>% select(Phylum, Family) %>% .[!duplicated(.$Family),]
# Bacteroidetes Bacteroidaceae
# Cyanobacteria Nostocaceae
# Proteobacteria Neisseriaceae
# Firmicutes Ruminococcaceae
# Actinobacteria ACK-M1
# Bacteroidetes Flavobacteriaceae
# Proteobacteria Pasteurellaceae
# Firmicutes Lachnospiraceae
# Proteobacteria Enterobacteriaceae
# Firmicutes Streptococcaceae
# 수동으로 순서 지정하기
Top10_SortByph <- c(# Actinobacteria
"ACK-M1",
"Other Actinobacteria",
# Bacteroidetes
"Bacteroidaceae", "Flavobacteriaceae",
"Other Bacteroidetes",
# Cyanobacteria
"Nostocaceae",
"Other Cyanobacteria",
# Firmicutes
"Ruminococcaceae", "Lachnospiraceae", "Streptococcaceae",
"Other Firmicutes",
# Proteobacteria
"Neisseriaceae", "Pasteurellaceae", "Enterobacteriaceae",
"Other Proteobacteria",
"Others")
◼ Others 처리
- 각 Phylum에서 top10을 제외한 family를 Other로 바꾸어 주기
Global.melt2$Family[Global.melt2$Phylum=="Actinobacteria" &
!Global.melt2$Family %in% c('ACK-M1')] <- "Other Actinobacteria"
Global.melt2$Family[Global.melt2$Phylum=="Bacteroidetes" &
!Global.melt2$Family %in% c( "Bacteroidaceae", "Flavobacteriaceae")] <- "Other Bacteroidetes"
Global.melt2$Family[Global.melt2$Phylum=="Cyanobacteria" &
!Global.melt2$Family %in% c( "Nostocaceae")] <- "Other Cyanobacteria"
Global.melt2$Family[Global.melt2$Phylum=="Firmicutes" &
!Global.melt2$Family %in% c("Ruminococcaceae", "Lachnospiraceae", "Streptococcaceae")] <- "Other Firmicutes"
Global.melt2$Family[Global.melt2$Phylum=="Proteobacteria" &
!Global.melt2$Family %in% c("Neisseriaceae", "Pasteurellaceae", "Enterobacteriaceae")] <- "Other Proteobacteria"
# 아까 지정한 family 순서 적용하기 (factor 이용)
Global.melt2$Family <- factor(Global.melt2$Family, levels = Top10_SortByph)
unique(Global.melt2$Family) # 16 level
# [1] Bacteroidaceae Nostocaceae Neisseriaceae Ruminococcaceae ACK-M1
# [6] Flavobacteriaceae Pasteurellaceae Lachnospiraceae <NA> Enterobacteriaceae
# [11] Other Proteobacteria Streptococcaceae Other Bacteroidetes Other Actinobacteria Other Firmicutes
# [16] Other Cyanobacteria
# 16 Levels: ACK-M1 Other Actinobacteria Bacteroidaceae Flavobacteriaceae Other Bacteroidetes ... Others
◼ 수동으로 색 지정하기
col <- c("#E78AC3","#E74AC3",
brewer.pal(3, "Blues"),
"#FDAE6B", "#E6550D",
brewer.pal(4, "Greens"),
brewer.pal(4, "BuPu"),
"#BDBDBD")- 각 Phylum별로 색을 다르게 지정하였으며, RColorBrewer 패키지를 사용하여 색이 잘 구분되게 그렸다.
◼ bar plot
p <- ggplot(data=Global.melt2, aes(x=Sample, y=Abundance, fill=Family)) +
geom_bar(aes(), position="fill", stat="identity") +
scale_fill_manual(values = col) +
theme_classic() +
theme(legend.position="bottom" ,
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_blank()) +
labs(y = "Relative Abundance (%)") +
theme(axis.text.x.bottom = element_text(angle = 45, vjust = 1, hjust=1),
axis.text = element_text(size = 7))
p
- 실수로 NA값을 처리하지 못했지만, family와 phylum level을 모두 나타내는 그림이다.
- 그러나 Phylum level을 모두 나타내지는 못했다 이는 Others에 포함되기 때문이다.
◼ 장점
- 가장 뚜렷한 색 구분으로 보는 이에게 확실한 의미를 전달할 수 있다
◼ 단점
- 손으로 수정해야 하는 부분이 가장 많음으로 번거로울 수 있다. (매우 귀찮다)
🟦 2. Phylum에 따라 색 나누는 hue_pal 함수 이용
◼ 패키지와 데이터 불러오기
library(phyloseq)
library(ggplot2)
library(dplyr)
library(RColorBrewer)
data("GlobalPatterns")
GlobalPatterns
df <- psmelt(GlobalPatterns)◼ Others 처리하기
df1 <- df
phylums <- c('Proteobacteria','Bacteroidetes','Firmicutes')
df1$Phylum[!df1$Phylum %in% phylums] <- "Others"
df1$Family[!df1$Phylum %in% phylums] <- "Others"
df1$Family[df1$Phylum=="Proteobacteria" &
!df1$Family %in% c('Alcaligenaceae','Enterobacteriaceae')] <- "Other Protobacteria"
df1$Family[df1$Phylum=="Bacteroidetes" &
!df1$Family %in% c('Bacteroidaceae','Rikenellaceae','Porphyromonadaceae')] <- "Other Bacteroidetes"
df1$Family[df1$Phylum=="Firmicutes" &
!df1$Family %in% c('Lactobacillaceae','Clostridiaceae','Ruminococcaceae','Lachnospiraceae')] <- "Other Firmicutes"
◼ 색을 지정해 줄 Phylum과 Family 추출하기
library(forcats)
library(dplyr)
df2 <- select(df1, Sample, Phylum, Family) %>%
mutate(Phylum=factor(Phylum, levels=c(phylums, "Others")),
Family=fct_reorder(Family, 10*as.integer(Phylum) + grepl("Others", Family))) %>%
group_by(Family) %>% # For this dataset only
sample_n(100) # Otherwise, unnecessary
df2
# Sample Phylum Family
# TRRsed3 Proteobacteria Alcaligenaceae
# TRRsed3 Proteobacteria Alcaligenaceae
# TRRsed3 Proteobacteria Alcaligenaceae
# TRRsed1 Proteobacteria Alcaligenaceae
# F21Plmr Proteobacteria Alcaligenaceae
# SV1 Proteobacteria Alcaligenaceae
# NP2 Proteobacteria Alcaligenaceae
# Even2 Proteobacteria Alcaligenaceae
# CL3 Proteobacteria Alcaligenaceae
# M31Fcsw Proteobacteria Alcaligenaceae
◼ 색 할당하기
# 출처 : https://stackoverflow.com/questions/49818271/stacked-barplot-with-colour-gradients-for-each-bar?rq=1
ColourPalleteMulti <- function(df, group, subgroup){
# Find how many colour categories to create and the number of colours in each
categories <- aggregate(as.formula(paste(subgroup, group, sep="~" )), df, function(x) length(unique(x)))
category.start <- (scales::hue_pal(l = 100)(nrow(categories))) # Set the top of the colour pallete
category.end <- (scales::hue_pal(l = 40)(nrow(categories))) # set the bottom
# Build Colour pallette
colours <- unlist(lapply(1:nrow(categories),
function(i){
colorRampPalette(colors = c(category.start[i], category.end[i]))(categories[i,2])}))
return(colours)
}
colours <- ColourPalleteMulti(df2, "Phylum", "Family")
◼시각화하기
library(ggplot2)
ggplot(df2, aes(x=Sample, fill = Family)) +
geom_bar(position="fill", colour = "grey") + # Stacked 100% barplot
scale_fill_manual("", values=colours) +
theme(axis.text.x=element_text(angle=90, vjust=0.5)) + # Vertical x-axis tick labels
scale_y_continuous(labels = scales::percent_format()) +
labs(y="Relative abundance")◼ 장점
- 각 Phylum 단계의 Others 도 포함하고 있어서 완전한 Phylum과 family의 정보까지 보여주는 그림이라고 할 수 있다.
◼ 단점
- Phylum에서 Others에 해당하는 것을 일일이 지정해 주어야 한다.
- 같은 Phylum에 여러 family가 있으면 색의 구분이 어려울 것이다.
🟦 3. fantaxtic package
- 🏠 : https://github.com/gmteunisse/Fantaxtic
- Phyloseq 객채에서 Taxonomy plot 그림에 전문화된 package로 손쉽게 원하는 taxa에 색 gradient를 줄 수 있다.
◼ Install
devtools::install_github("gmteunisse/fantaxtic")
require("fantaxtic")
require("phyloseq")
library(tidyverse)◼ 색을 지정할 Taxa level 결정
top_nested <- nested_top_taxa(GlobalPatterns,
top_tax_level = "Phylum",
nested_tax_level = "Genus",
n_top_taxa = 3,
n_nested_taxa = 3)- top_tax_level = "Phylum" # 메인 색을 정할 top taxa level
- nested_tax_level = "Genus" # 색의 변화로 표현할 nested taxa level
n_top_taxa = 3, # top taxa level의 수
n_nested_taxa = 3 # nested taxa level의 수
◼ Phylum의 total abundance순으로 정렬
- 위 예시에서는 Proteobacteria를 기준을 정렬하겠다.
# Order samples by the total abundance of Proteobacteria
sample_order <- psmelt(top_nested$ps_obj) %>%
data.frame() %>%
# relative abundances 계산
group_by(Sample) %>%
mutate(Abundance = Abundance / sum(Abundance)) %>%
# 원하는 Phylum level로 정렬한다
filter(Phylum == "Proteobacteria") %>%
group_by(Sample) %>%
summarise(Abundance = sum(Abundance)) %>%
arrange(Abundance) %>%
# Extract the sample order
pull(Sample) %>% as.character()
sample_order
# [1] "TS29" "TS28" "M31Fcsw" "M11Fcsw" "LMEpi24M" "AQC1cm" "AQC4cm" "SLEpi20M"
# [9] "Even2" "Even3" "Even1" "M11Tong" "M31Plmr" "AQC7cm" "F21Plmr" "NP5"
# [17] "CC1" "SV1" "CL3" "NP3" "NP2" "M11Plmr" "TRRsed1" "M31Tong"
# [25] "TRRsed3" "TRRsed2"
◼ 시각화
# Plot
p <- plot_nested_bar(top_nested$ps_obj,
"Phylum",
"Genus",
sample_order = sample_order,
nested_merged_label = "NA and other")
p + labs(title = "Relative abundances of the top 3 species for each of the top 3 phyla")
p + theme(panel.background = element_rect(fill='transparent'),
plot.background = element_rect(fill='transparent', color=NA),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.text.x = element_text(hjust = 1, vjust = 1, angle = 45, size = 10)
)
p + facet_wrap(~SampleType, scales = "free_x")이렇게 facet_wrap()을 이용해 SampleType별로도 관찰이 가능하다
◼ 장점
- 가장 간단한 코드로 깔끔한 그림을 얻을 수 있다.
- Top taxa를 여러 taxa 레벨로 나누어서 색을 자동 지정해 주기 때문에 시각화 시 매우 편리하다.
◼ 단점
- 실제로 사용해 본 결과, 각 Phylum에 색이 5가지 이상이면 ggnested함수가 이를 인식하고, 위 패키지의 기본 색이 부여되는데, 이 색도 2번 방법처럼 각 taxa level마다 정확한 색 구분이 어렵다.
- 또한 ggnested 함수로 넘어가면 Phylum와 Genus 레벨을 보여주는 것이 아닌, Genus level만 보여주고 있지만, index의 카테고리는 Phylum level이 적혀있다. 이는 보는 이로 하여금 위 그래프가 Phylum 레벨의 내용을 모두 설명하는 것처럼 보일 수도 있다.
- 또한 index 간의 위치를 바꾸는 것이 어렵고, Phylum 레벨에서만 바꾸는 것이 가능하다.
🟦 결론
◼ 편한 방법만이 좋은 것은 아니다.
- 3번째 패키지가 가장 사용하기 편리하지만, 생각보다 제한이 많아서 자주 사용하게 되지는 않는 것 같다.
- 수동으로 일일이 지정하는 것은 귀찮지만 함수화 하여 필요할 때마다 불러오는 것이 훨씬 효과적일 것이다. 초기에 시간을 들여 사용한 함수들은 나중에 시간활용 부분에서 큰 도움이 된다. 그러므로 다시 만들러 가자..ㅠ
🟦 Reference
- https://github.com/gmteunisse/Fantaxtic