
작성: 2023.10.13
논문을 쓸 때, 특정 taxa에 대한 구체적인 abundance값을 기입하는 경우가 많습니다. 보통 "mean: 38.9%, SD 4.3 %"의 형태로 많이 작성되는데요, 이 값들은 어떻게 구하는 것이 좋을까요?
1. 먼저 가장 기본적인 엑셀을 활용할 수 있습니다.
- OTU와 taxonomy table을 기반으로 작성된 abundance값으로 함수를 입력하여 구하는 것입니다.
- 가장 많이 사용되는 방법이라고 볼 수 있습니다.
2. R의 data.frame형태에서 계산할 수 있습니다.
- 약간의 코딩이 필요하지만 chatGPT가 도와줄 것입니다.
- R이 익숙한 분이라면, 엑셀보다 훨씬 간단하게 구할 수 있을 것입니다.
- 또한 함수로 만들어 놓으면, 다른 샘플에서도 더 간편하게 사용할 수 있습니다.
우리가 코딩을 하는 이유는 효율성에 있습니다. 지금의 시간 투자가 미래의 시간 낭비를 줄여줍니다.
지난 글(Alpha diveristy의 Mean, Sd, N, Upper, Lower, 95% CI값과 p-value값 저장)을 참고하여 만들어 봅시다.
이번 글에는 예제 샘플도 추가하였습니다.
예제 데이터
- QIIME2 tutorial인 moving-picture의 데이터입니다. 사람의 양 손바닥, 혀, 장의 마이크로바이옴 데이터를 담고 있습니다. 현재 저장된 형식은 phyloseq object입니다. phylsoeq형식은 biom format을 차용하여 R에서 편리하게 다루도록 개발된 것으로, metadata와 otu table, taxonomy 그리고 계통정보를 한 번에 저장할 수 있습니다.
- 우리는 이 body.site에 따른 마이크로바이옴의 상대적 풍부도의 통계값을 관찰해 봅시다.
코드
먼저 파일을 불러옵니다.
library(dplyr)
library(phyloseq)
library(rlang)
ps <- readRDS("./ps.rds")
ps.rel <- phyloseq::transform_sample_counts(ps, fun = function(x) x/sum(x))
이제 계산 함수를 만듭니다.
Abund_cal <- function(ps.glom, tax_level, group, path) {
# melt
melt <- psmelt(ps.glom)
# setting
Taxonomy <- as.vector(unique(melt[, tax_level]))
meta <- data.frame(sample_data(ps.glom))
meta_com <- meta[, group, drop = TRUE] %>% unique
tax_num <- length(Taxonomy)
# reset
Total_result <- NULL
## Total abundance calculation
Total_result <- melt %>%
dplyr::group_by(!!rlang::sym(tax_level)) %>%
dplyr::summarize(
Total.Mean = mean(Abundance), # 평균
Total.N = n(), # 행 개수
Total.Sd = sd(Abundance) # 표준편차
)
## Each group abundance calculation
for (com in meta_com) {
# 각 그룹별 계산
melt.2 <- melt %>% filter(!!rlang::sym(group) == com)
result <- melt.2 %>%
dplyr::group_by(!!rlang::sym(tax_level)) %>%
dplyr::summarize(
!!paste0(com, ".Mean") := mean(Abundance), # 평균
!!paste0(com, ".N") := n(), # 행 개수
!!paste0(com, ".Sd") := sd(Abundance) # 표준편차
) %>%
dplyr::mutate(!!paste0(com, ".se") := !!rlang::sym(paste0(com, ".Sd")) / sqrt(!!rlang::sym(paste0(com, ".N"))), # 표준오차
!!paste0(com, ".lower") := !!rlang::sym(paste0(com, ".Mean")) - qnorm(0.975) * !!rlang::sym(paste0(com, ".se")), # 95% 신뢰 구간 하한
!!paste0(com, ".upper") := !!rlang::sym(paste0(com, ".Mean")) + qnorm(0.975) * !!rlang::sym(paste0(com, ".se")), # 95% 신뢰 구간 상한
!!paste0(com, ".CI95per") := !!rlang::sym(paste0(com, ".upper")) - !!rlang::sym(paste0(com, ".lower")) # 95% 신뢰 구간
)
# abundance 결과 합치기
Total_result <- bind_cols(Total_result, result[, -1])
}
print(Total_result)
write.csv(Total_result, paste0(path, "Taxa_", group, "_stat_", tax_level, ".csv"), row.names = F)
}
이 계산함수를 활용하여 2가지 결과물을 만들어 봅니다.
ps.rel.ph <- tax_glom(ps.rel, "Phylum")
ps.rel.gn <- tax_glom(ps.rel, "Genus")
Abund_cal(ps.glom = ps.rel.ph, tax_level = "Phylum", group = "body.site", path = "./")
Abund_cal(ps.glom = ps.rel.gn, tax_level = "Genus", group = "body.site", path = "./")
결과물
실제 저장된 결과물은 아래에서 확인할 수 있습니다.
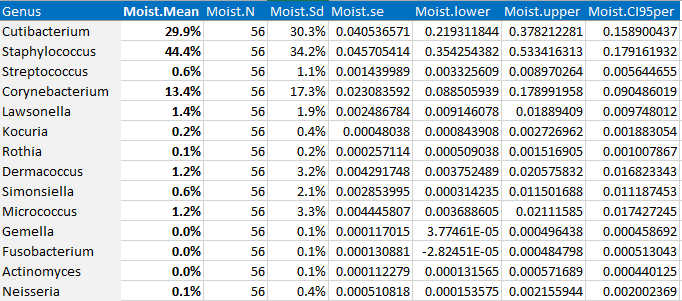
Phylum level에서 살펴볼까요? (결과물은 눈에 잘 보이도록 엑셀에서 수정)
- 먼저 Mean 값은 각 타입에 따른 평균 abundance를 뜻합니다. 제가 이 부분을 엑셀에서 %로 바꾸어놓았습니다.
- 제일 왼쪽 열에는 위 샘플에 존재하는 모든 Phylum을 관찰할 수 있습니다.
- Total.열에서는 전체 샘플에 해당하는 평균 abundance와 샘플 수(N)와 표준편차를 구했습니다.
- 그 오른쪽을 보시면 각 부위 (gut, left.palm, right.palm, toungue)에 따른 abudance의 통계값이 위치하고 있습니다.

자 이제 논문을 적을때, "Bacteriodetes는 전체 34개의 샘플에서 26.2%로 존재하며, 특히 Gut에서 우세하다(mean: 34.78%, SD 5.1%)"라고 적을 수 있습니다.
지난글을 참고하여, 각 Genus의 풍부도에 따른 p-value값만 가진 표도 제작할 수 있을 것입니다.
이렇게 한 번 만들어 놓으면 다음 연구에서는 논문을 적을 때 훨씬 수월하겠죠?
읽어주셔서 감사합니다.