
작성 2023.09.04
수정 2023.09.26

🟩 Vegan
- biplot이란 하나의 그림에 두 개의 데이터를 보여주는 plot이다.
- Vegan 패키지는 환경데이터를 처리 및 분석에 사용된다. 마이크로바이옴 데이터와 환경데이터는 샘플이름이 열에, 환경 또는 미생물의 이름이 행에 위치(혹은 그 반대)하는 feature table을 분석에 이용하기 때문에, 많은 분석 방법을 공유한다.
- 이 중에서 vegan의 envfit 함수를 이용한 biplot을 phyloseq object를 사용해 그려보자.
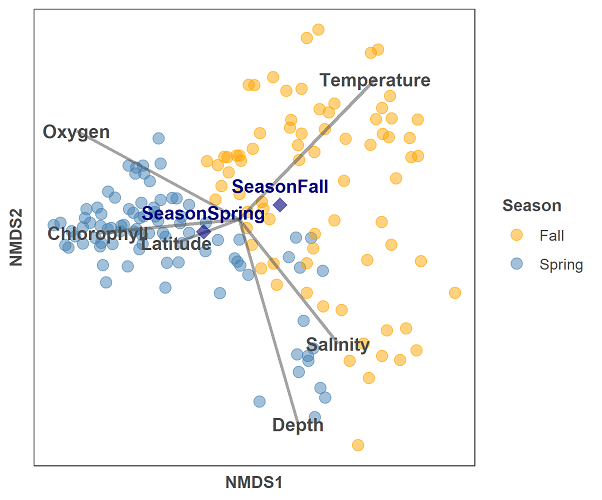
- 위처럼 샘플을 point로 나타내고, 관련 메타데이터를 arrow로 그리거나, feature(ASV)를 arrow로 표시하는 경우가 있다.
🟩 Example data
- QIIME2 tutorial (moving-picture)
- Human microbiome
🟩 Code
library(phyloseq)
library(ggplot2)
library(vegan)
library(dplyr)
library(tibble)
ps <- readRDS("~/ps.rds")
ps.rel <- phyloseq::transform_sample_counts(ps, fun = function(x) x/sum(x))
1. tax plot and sample arrow
- NMDS plot
- point : Class
- arrow : sampleID
# Class를 기준으로 otu table합치기
ps.cl <- tax_glom(ps.rel, taxrank = "Class")
# phyloseq에서 otu, taxonomy 추출
otu <- otu_table(ps.cl) %>% t()
tax <- tax_table(ps.cl) %>% as.matrix()
#bray-curtis matrix를 사용한 taxonomy 간의 거리계산
dist <- phyloseq::distance(ps.cl, method = "bray", type = "species") # type = "samples"였다면 샘플 기준으로 거리 측정
set.seed(42)
ord <- phyloseq::ordinate(physeq = ps.cl, method = "NMDS", distance = dist) # NMDS plot 생성
NMDS = data.frame(NMDS1 = ord$points[,1], NMDS2 = ord$points[,2]) # NMDS1, 2 dimension 추출
NMDS.2 <- merge(NMDS, as.data.frame(tax[, "Phylum"]), by = "row.names") # Class 라벨링
# envfit 계산
env <- envfit(NMDS, t(otu), perm = 999)
vec<-as.data.frame(env$vectors$arrows) # vector 성분 추출
vec.2 <- cbind(vec, `SampleID` =rownames(vec)) # SampleID로 라벨링
# envfit을 사용한 plot 그리기
p <- ggplot(NMDS.2) +
geom_point(mapping = aes(x = NMDS1, y = NMDS2, color = Phylum), alpha = 0.7) + # 색은 Phylum기준으로
coord_fixed() +
geom_segment(data = vec.2,
aes(x = 0, xend = NMDS1, y = 0, yend = NMDS2),
arrow = arrow(length = unit(0.25, "cm")), colour = "grey") +
geom_text(data = vec.2, aes(x = NMDS1, y = NMDS2, label = SampleID),
size = 3) +
theme_bw()
p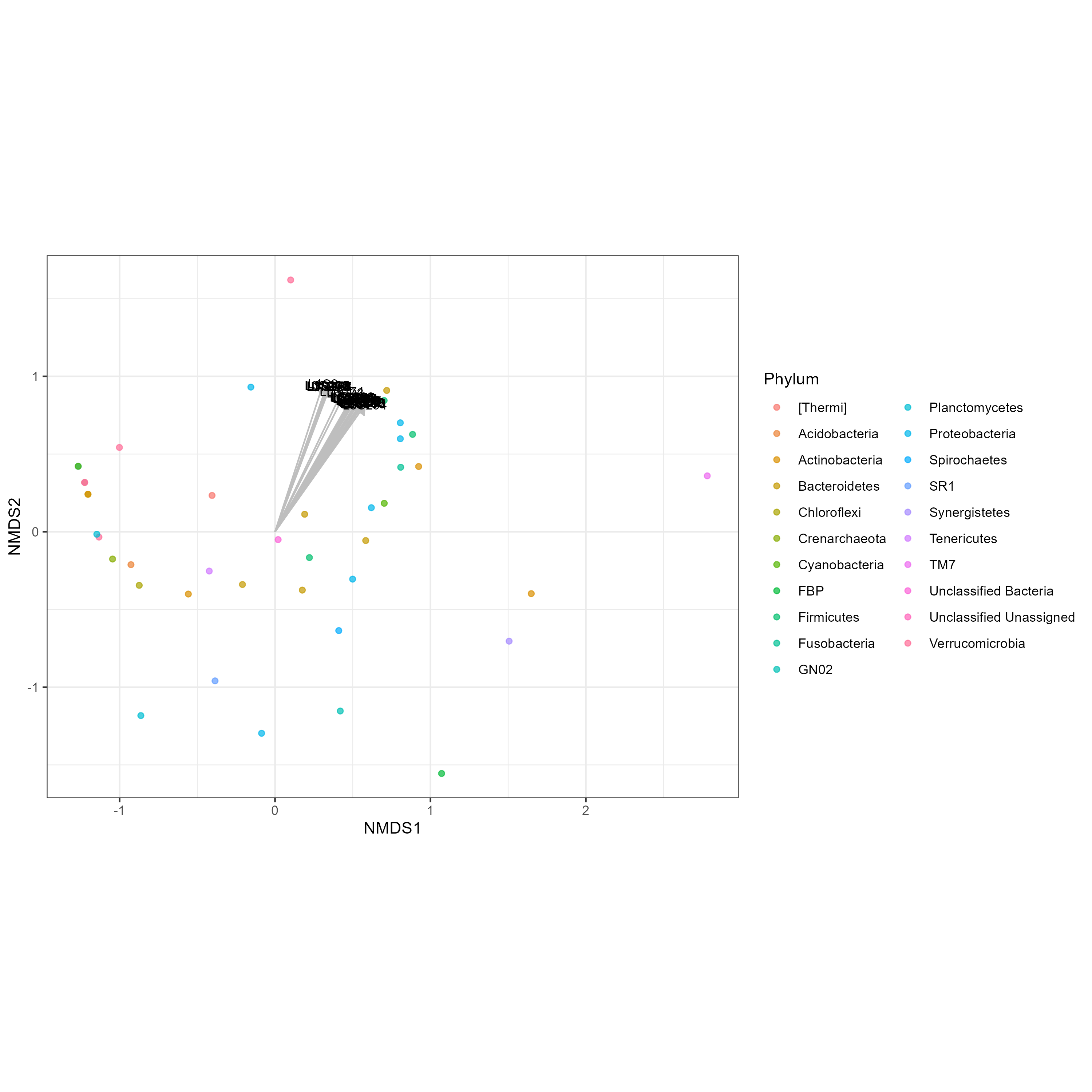
2. sample plot and taxa arrow
- NMDS plot
- point : sampleID
- arrow : Phylum
ps.g <- ps.rel %>% tax_glom(taxrank = "Phylum")
otu <- otu_table(ps.g)
tax <- tax_table(ps.g) %>% as.matrix()
s.dist <- phyloseq::distance(ps.g, method = "unifrac", type = "Samples")
set.seed(42)
s.ord <- phyloseq::ordinate(physeq = ps.g, method = "NMDS", distance = s.dist)
s.NMDS = data.frame(NMDS1 = s.ord$points[,1], NMDS2 = s.ord$points[,2])
s.NMDS.2 <- merge(s.NMDS, sample_data(ps.g)[, "body.site"], by = "row.names")%>%
column_to_rownames("Row.names")
s.env <- envfit(s.NMDS, t(otu), perm = 999)
s.env
ordiplot (s.ord, display = 'sites')
plot (s.env)
하지만 여기서 p-value값이 0.05가 넘지 않는 taxa를 제외한 후 관찰해 보자.
s.vec<-as.data.frame(s.env$vectors$arrows)
s.vec$p.val <- s.env$vectors$pvals
s.vec2 <- s.vec[s.vec$p.val < 0.05, ]
s.vec3 <- merge(s.vec2, tax[, "Phylum"], by = "row.names") %>%
column_to_rownames("Row.names")
p <- ggplot(s.NMDS.2) +
geom_point(mapping = aes(x = NMDS1, y = NMDS2, color = body.site), alpha = 0.7) +
coord_fixed() + ## need aspect ratio of 1!
geom_segment(data = s.vec3,
aes(x = 0, xend = NMDS1, y = 0, yend = NMDS2),
arrow = arrow(length = unit(0.25, "cm")), colour = "grey") +
geom_text(data = s.vec3, aes(x = NMDS1, y = NMDS2, label = Phylum), size = 3, check_overlap=TRUE) +
# check_overlap=TRUE : 중복된 label제거
theme_bw()
p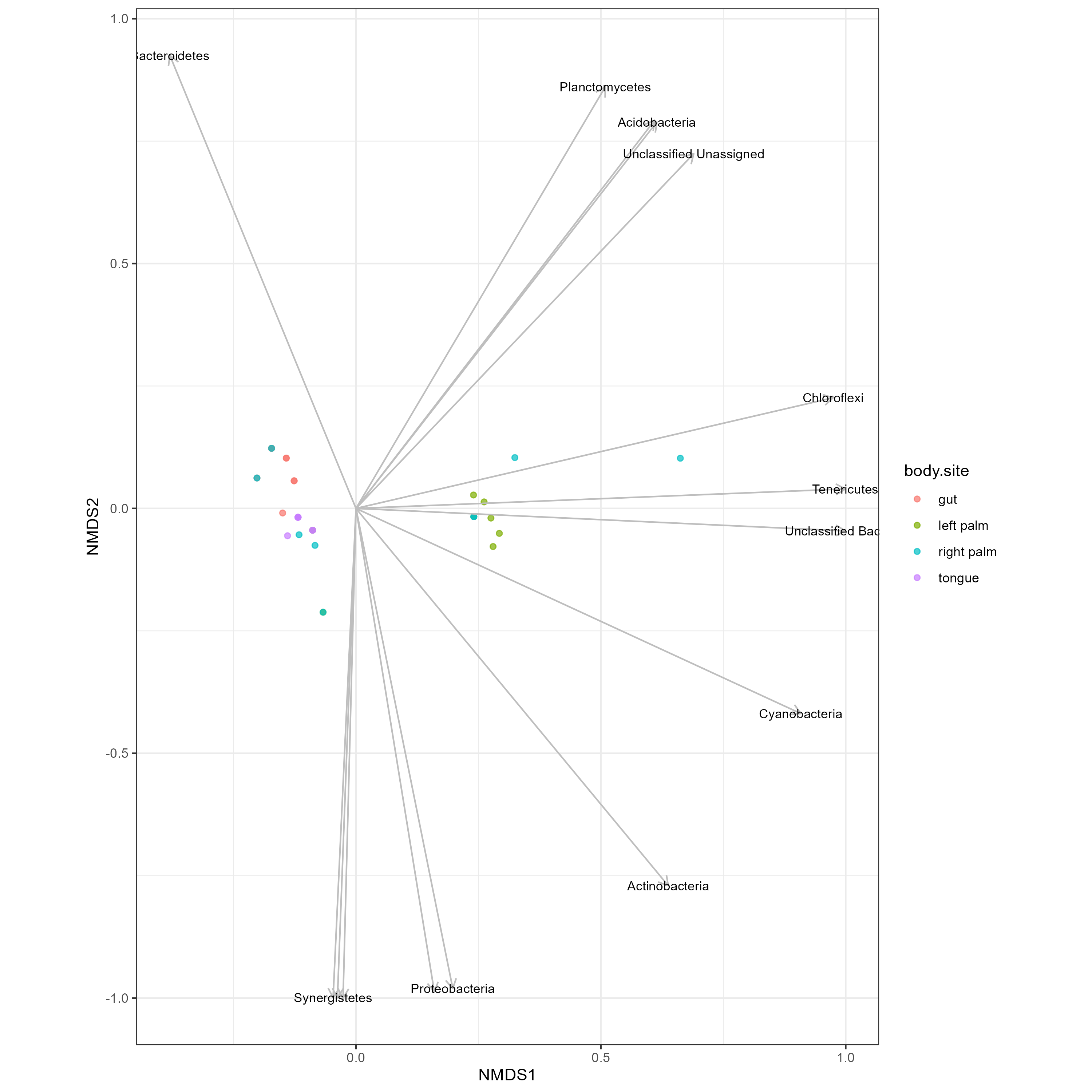
3-1. sample plot and taxa arrow + PCoA
- PCoA plot
- point : sampleID
- arrow : Phylum
# phyloseq에서 데이터 추출
ps.g <- ps.rel %>% tax_glom(taxrank = "Phylum")
otu <- otu_table(ps.g) %>% as.matrix()
tax <- tax_table(ps.g) %>% as.matrix()
groups <- sample_data(ps.g)[, "body.site"]
# 샘플간 거리 구하기
s.dist <- phyloseq::distance(ps.g, method = "wunifrac", type = "Samples")
# PCoA 축 구하기
pcoa <- cmdscale(s.dist)
# envfit계산
efit <- envfit(pcoa, t(otu_table(ps.g)))
# p-val 보정
ef.adj <- efit
pvals.adj <- p.adjust (efit$vectors$pvals, method = 'bonferroni')
ef.adj$vectors$pvals <- pvals.adj
ef.adj
# arrow 거리 보정
ef.arr<-as.data.frame(ef.adj$vectors$arrows*sqrt(ef.adj$vectors$r))
# p-val 0.05이하만 추출
s.vec <- ef.arr
s.vec$p.val <- ef.adj$vectors$pvals
s.vec.2 <- s.vec[s.vec$p.val < 0.05, ]
s.vec.3 <- merge(s.vec.2, tax[, "Phylum"], by = "row.names") %>% column_to_rownames("Row.names")
# pcoa와 metadata 합치기
pcoa.2 <- cbind(pcoa, groups)
# plot
p <- ggplot(pcoa.2) +
geom_point(aes(x=`1`, y=`2`, color = body.site), alpha = 0.7) +
coord_fixed() +
geom_segment(data = s.vec.3,
aes(x = 0, xend = Dim1, y = 0, yend = Dim2),
arrow = arrow(length = unit(0.25, "cm")), colour = "grey") +
geom_text(data = s.vec.3, aes(x = Dim1, y = Dim2, label = Phylum), size = 3, check_overlap=TRUE) +
theme_bw()
p
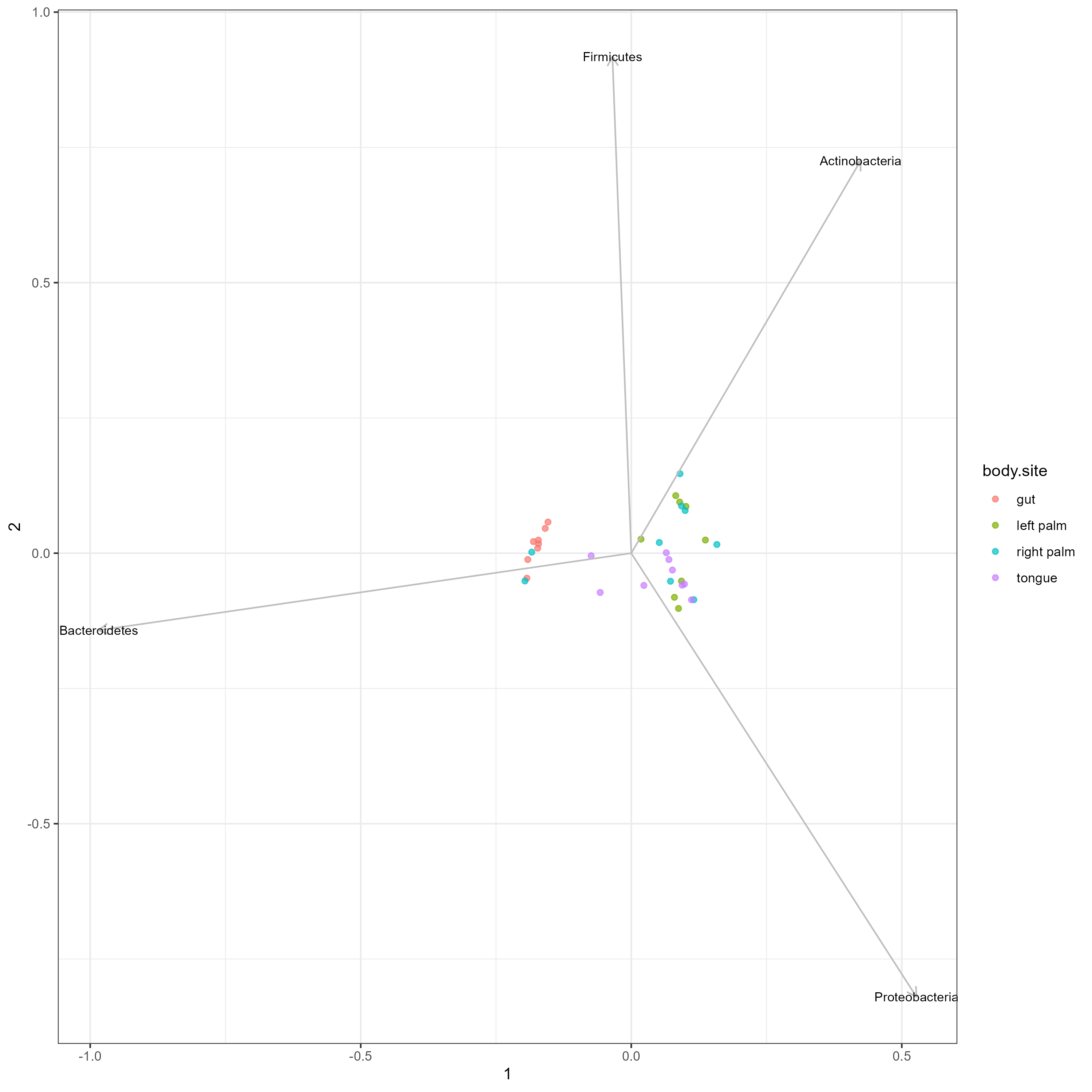
Phylum level을 표시하는 Arrow의 길이가 상당히 길게 잡혔다. 만약 Genus를 기준으로 살펴본다면 arrow의 선이 짧아질 것 같다.
arrow의 거리 보정은
ef.arr <-as.data.frame(ef.adj$vectors$arrows*sqrt(ef.adj$vectors$r))
코드로 적용된다. 본인의 result에 맞게 배수를 곱해줘서 적당하게 맞추어 보자.
3-2. sample plot and taxa arrow + PCoA
- PCoA plot
- point : sampleID (gut and tongue)
- arrow : Genus
ps.gn <- ps.rel %>% tax_glom(taxrank = "Genus") %>%
subset_samples(body.site %in% c("gut", "tongue"))
otu <- otu_table(ps.gn) %>% as.matrix()
tax <- tax_table(ps.gn) %>% as.matrix()
# 거리 구하기
s.dist <- phyloseq::distance(ps.gn, method = "wunifrac", type = "Samples")
groups <- sample_data(ps.gn)[, "body.site"]
PCoA <- phyloseq::ordinate(physeq = ps.gn, method = "PCoA", distance = s.dist)
pcoa <- PCoA$vectors[, 1:2]
envfit <- envfit(pcoa, t(otu_table(ps.gn)))
ef.arr<-as.data.frame(envfit$vectors$arrows*sqrt(envfit$vectors$r))
s.vec <- ef.arr
s.vec$p.val <- envfit$vectors$pvals
s.vec.2 <- s.vec[s.vec$p.val < 0.05, ]
s.vec.3 <- merge(s.vec.2, tax[, "Genus"], by = "row.names") %>% column_to_rownames("Row.names")
pcoa.2 <- cbind(pcoa, groups)
p <- ggplot(pcoa.2) +
geom_point(aes(x=`Axis.1`, y=`Axis.2`, color = body.site), alpha = 0.7) +
coord_fixed() +
geom_segment(data = s.vec.3,
aes(x = 0, xend = Axis.1, y = 0, yend = Axis.2),
arrow = arrow(length = unit(0.25, "cm")), colour = "grey") +
geom_text(data = s.vec.3, aes(x = Axis.1, y = Axis.2, label = Genus), size = 3, check_overlap=TRUE) +
theme_bw()
p- p-value값 보정하면 유의한 feature가 존재하지 않아서, 위 예제 데이터에서는 실행하지 않음
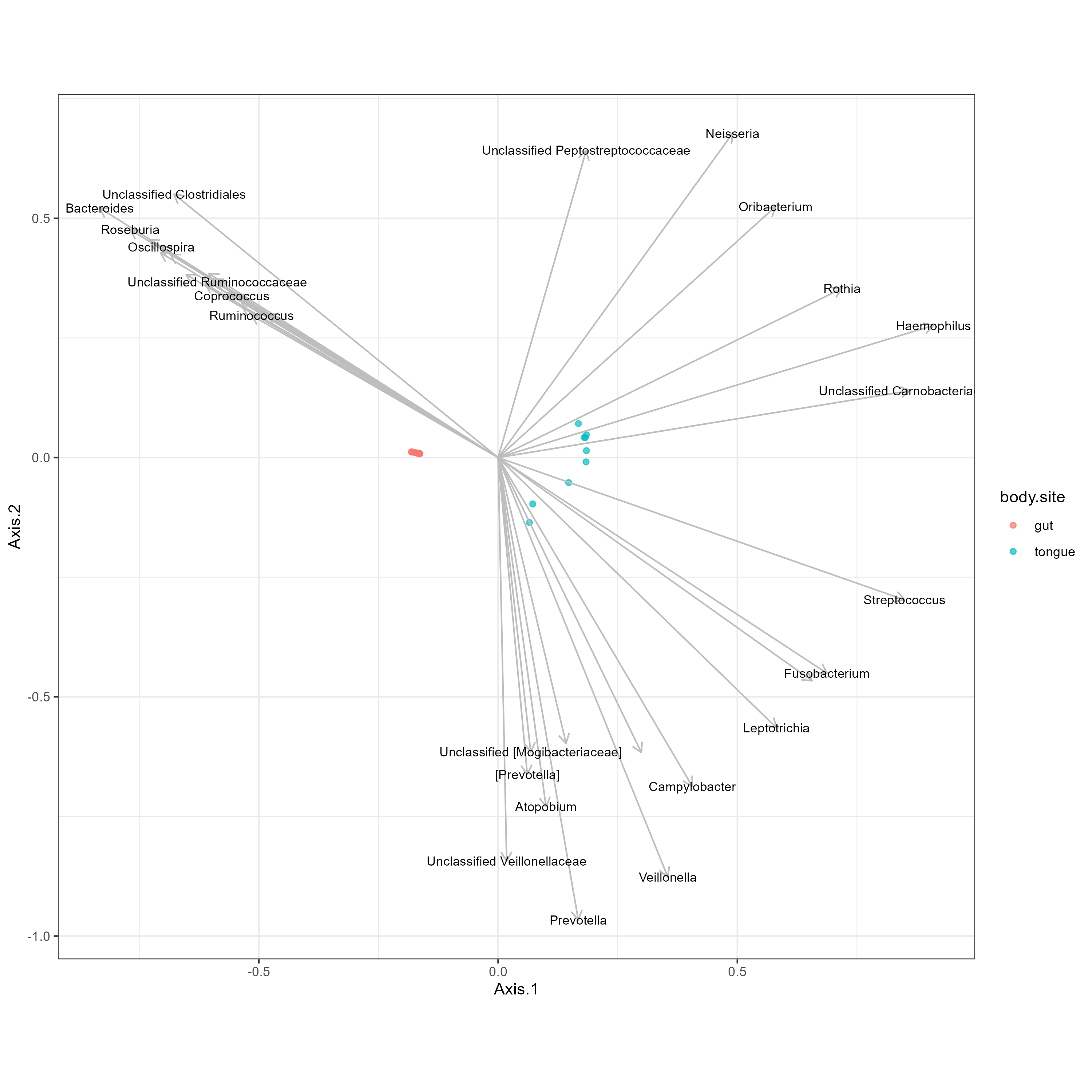
Gut에서 유의하게 많이 분포하는 Genus가 완벽하게 Gut 샘플 방향을 가리키고 있지 않지만, 경향을 가진다고 해석하면 될 듯하다.
3-3. 추가
만약 ASV에서 genus, phylum으로 데이터를 합치지 않고, 먼저 ASV상태에서 PCoA를 그린 다음, 화살표만 Genus나, Phylum으로 바꾸고 싶다면 아래 함수를 사용해서 원하는 taxa(아래는 Genus)를 기준으로 Axis1과 Axis2를 평균 내주면 된다.
그렇게 하면 원형의 Sample당 PCoA값이 변형되지 않게 PCoA를 그릴 수 있다.
s.vec.fdr.genus <- s.vec.3 %>%
group_by(Genus) %>%
summarize_all(.funs = list(mean)) %>%
select(Genus, Axis.1, Axis.2)
참고
- https://www.introranger.org/post/ord-groups-gradients/
- https://fromthebottomoftheheap.net/slides/intro-vegan-webinar-2020/intro-to-vegan.html#55
- https://www.davidzeleny.net/anadat-r/doku.php/en:suppl_vars_examples
- https://jkzorz.github.io/2020/04/04/NMDS-extras.html
- https://fukamilab.github.io/BIO202/06-A-unconstrained-ordination.html
아무리 뒤져봐도 phyloseq에서 바로 biplot을 그리는 함수는 없는 것 같다.