
- 작성 : 2023.08.29
1. MicrobiomeMarker 패키지에 대해서
- 논문 : Cao, Y., Dong, Q., Wang, D., Zhang, P., Liu, Y., & Niu, C. (2022). microbiomeMarker: an R/Bioconductor package for microbiome marker identification and visualization. Bioinformatics (Oxford, England), 38(16), 4027–4029. https://doi.org/10.1093/bioinformatics/btac438
- 인용수 : 34 (2023.08.29 기준)
- 공식 문서 : https://yiluheihei.github.io/microbiomeMarker/index.html
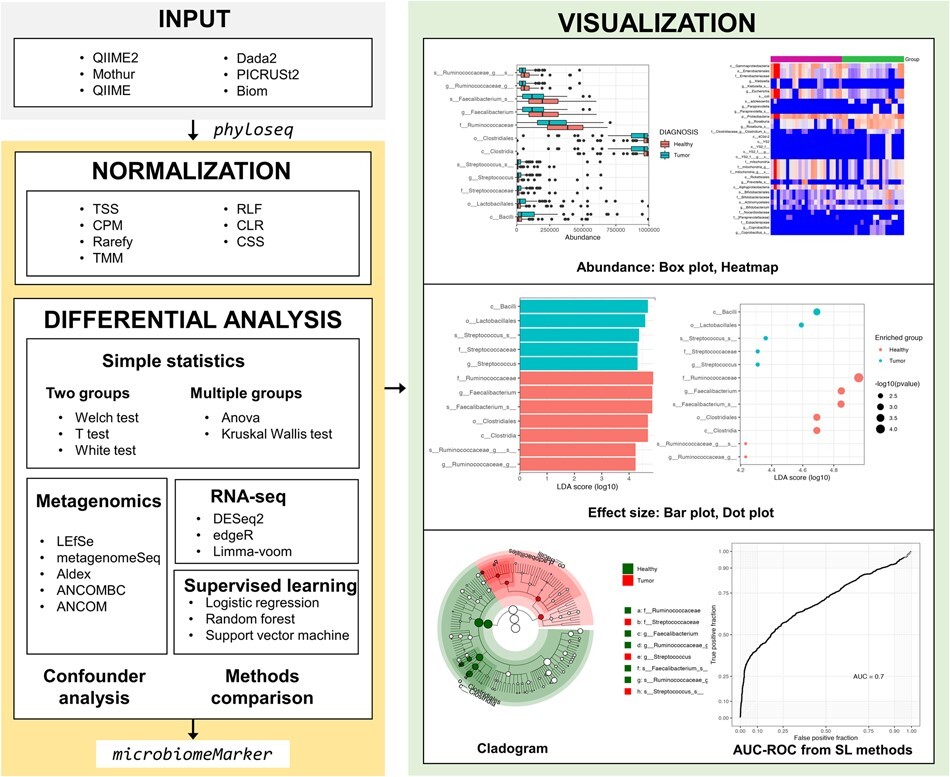
MicrobiomeMarker이란 여러 통계분석 기법이나 미생물 데이터 전용 차등분석을 이용하여 생물학적 marker를 구하는 R기반 패키지이다. 위 패키지는 여러 분석 패키지를 모아놓은 총집합 도구라고 할 수 있으며, 막연했던 머신러닝 기법까지 명령어 몇 줄로 실행할 수 있게 해 준다. 또한 Input 파일로는 phyloseq을 받기 때문에, 기존 phyloseq사용자들은 손쉽게 사용 가능하다.
MicrobiomeMarker가 지원하는 분석법은 아래와 같다.
- Two groups : whelch test, t-test, white test
- Multiple group : Anova, Kruskal wallis test
- Metagenome : LEfse, metagenomeSeq, ALDEx2, ABCOMBC, ANCOM
- RNA-seq : DESeq2, egdeR, Limma-voom
- Supervised learning : Logistic regression, Random forest, Support vector machine
분석뿐만 아니라 bar plot이나 dot plot을 통해 각 marker를 시각화해 주며, cladogram으로 계통수에 접목시켜 보여주기도 한다. 머신러닝의 경우 AUC-ROC curve를 통해 결괏값의 정확도를 설명해 준다.
2. 머신러닝이란?
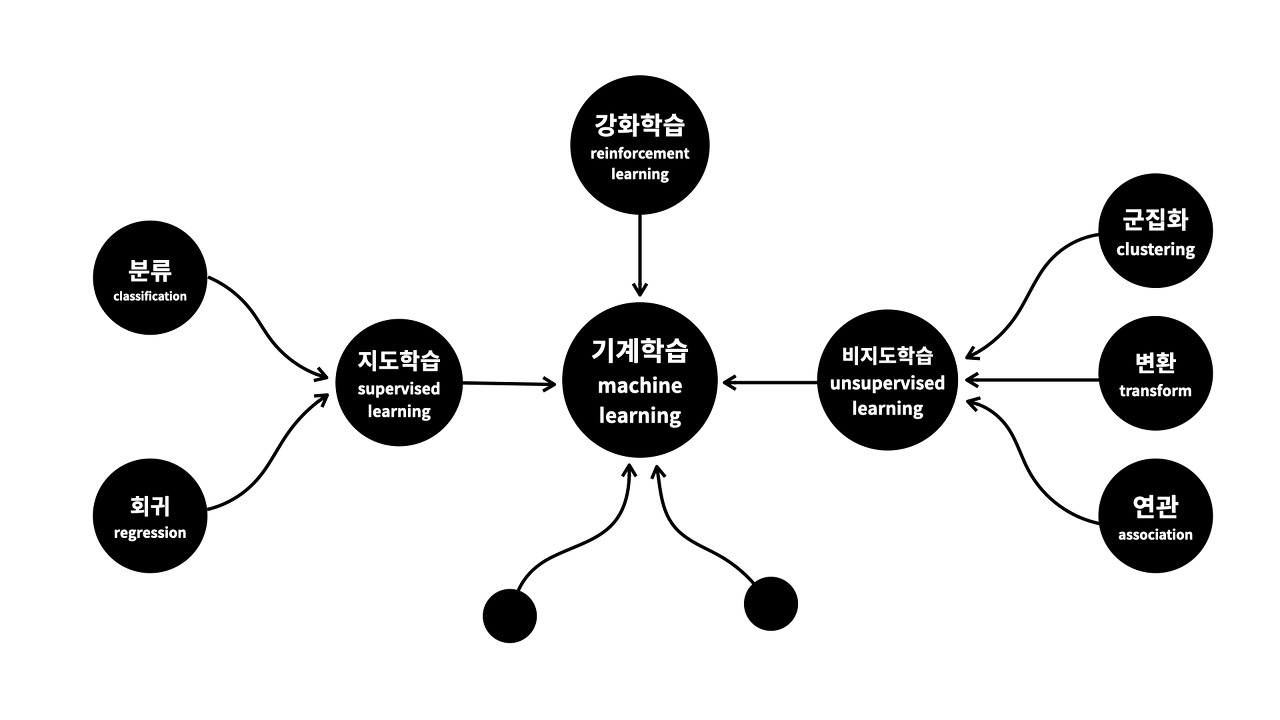
- 최근 AI, 머신러닝은 여러 연구에 접목되고 있다. 그러나 이는 모두 통계 기술 중 하나로 분류와 예측에 사용된다. 수학적 이해는 어렵지만, 활용법만 알면 사용하는 것은 쉽다.
- 분류는 어떤 그룹을 특정 기준으로 나누어 주는 것이며, 예측은 과거의 데이터로 미래의 데이터를 예측하는 것이다.
- 이때 사용되는 방법을 크게 지도학습, 비지도 학습으로 나누어 준다. 각 방법의 예시로 대략적인 모델 구동방식을 생각해 보자.
e.g.) 지도학습(분류) : 고양이와 개의 사진을 학습시키고, 어떠한 사진이 주어졌을 때 분류시키는 것
e.g.) 지도학습(예측) : 방문자수와 구매율 데이터를 학습시키고 내년의 매출을 예측시키는 것
e.g.) 비지도 학습(군집화) : 고양잇과 개의 사진을 10,000장을 주고 이를 두 그룹으로 나누라고 시킨다
e.g.) 비지도학습(변환-차원축소) : 마이크로바이옴 데이터를 relative abundance로 변환하거나, 각 샘플의 거리는 bray-curtis dissimilarity로 변환하는 것
e.g.) 비지도학습(연관) : A물건을 산 사람이 B물건을 살 확률은 무엇인가?
3. 마이크로바이옴 데이터에서 머신러닝(ML) 방법의 적용이란?
많은 사람들이 마이크로바이옴 분석을 위해 일상적으로 머신러닝 기법을 사용하고 있다.
1. Feature(OTU, ASV) inference
- qiime에서 기본적인 feature를 구하는 방법은 Naïve Bayes classifiers를 사용해 분류된다.
2. Diversity
- Beta diversity 분석에서 PCA, PCoA를 통해 차원축소를 수행한다.
3. Biomarker 구하기
- 가장 많이 사용되었던 microbiomeMarker를 발굴하는 LEfSe에서 사용되는 Linear discriminant analysis (LDA)도 ML방법 중 하나이다.
4. Supervised leaning방법으로 maker microbiome 구해보기
- MicrobiomeMarker 패키지에서 지원하는 sl분석 방법은 Logistic regression(LR), Random forest(RF), Support vector machine(SVM)이 있다. 이는 지도학습으로 기존 데이터를 학습시킨 후 적용한다.
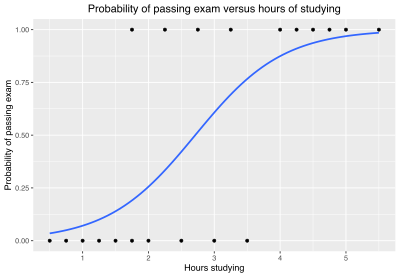
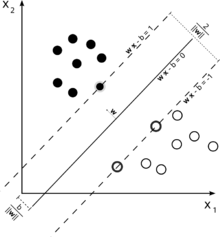
1) Logistic regression란 기존의 선형적인 데이터를 분석하는 것이 아니라 범주형 데이터(남, 여/ 0 또는 1)를 분석하는 방법이다. 즉 아래 그림과 같이 y값이 0과 1 만들 가지고 있지만 이를 예측하고 분류해 내는 분석법을 말한다.
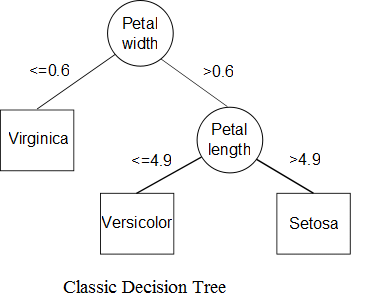
2) Random forest란 여러 결정트리를 학습(bagging)하는 앙상블(여러 분석의 총집합) 분석 방법이다.
- 트리가 여러 개니까 포레스트...라는 이름을 붙인 분석 방법이다(팩트).
- 트리는 의사결정트리를 말한다. R을 배울 때 가장 먼저 접하게 되는 iris데이터를 의사결정 트리로 분류해 보자. 이때 각 node는 샘플을 나누는 가장 큰 기준이다.
3) Support vector machine란 각 데이터를 가장 큰 거리로 나누는 선을 찾는 방법이다.
- 이때 두 범주 사이에 가장 가까운 샘플 간의 거리(w)가 최대가 되는 선(vector)을 구한다.
※ 예제 데이터는 샘플 수도 매우 적기 때문에 왜곡된 결과가 나올 수 있다는 점을 감안 ※
Import Data
- QIIME2 tutorial moving-picture data
- human body site : left and right palm, gut, tongue
- Input file : phyloseq object(Not relative abundance otu table)
library(phyloseq)
library(microbiomeMarker)
library(ggplot2)
library(dplyr)
ps <- readRDS("./ps.rds") %>%
subset_samples(body.site %in% c("gut", "tongue"))
ps
Logistic regression
Read count and all taxa
LR.1 <- microbiomeMarker::run_sl(
ps = ps,
group = "body.site",
method = "LR")
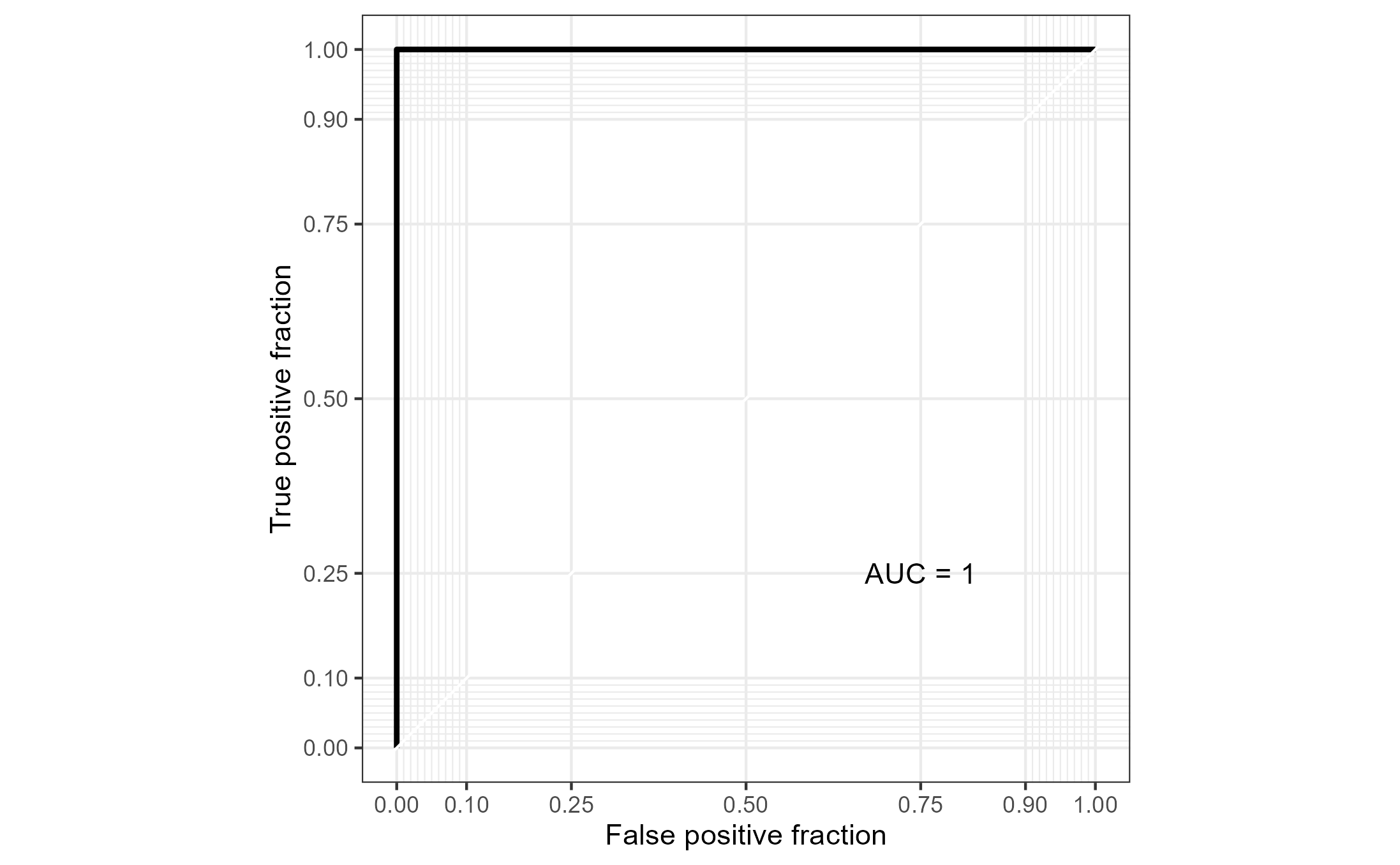
microbiomeMarker::plot_sl_roc(LR.1, group = "body.site")
microbiomeMarker::plot_ef_bar(LR.1)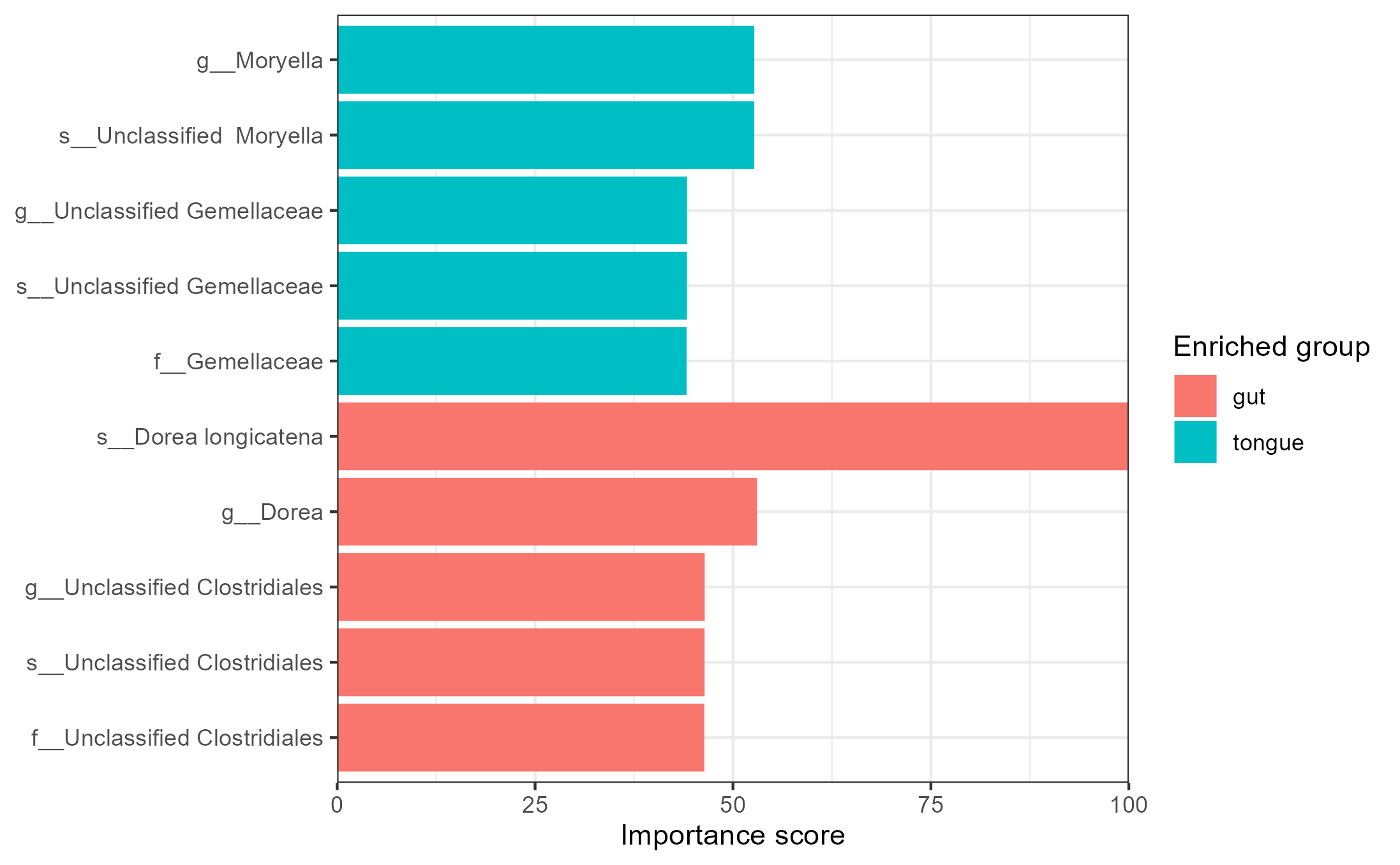
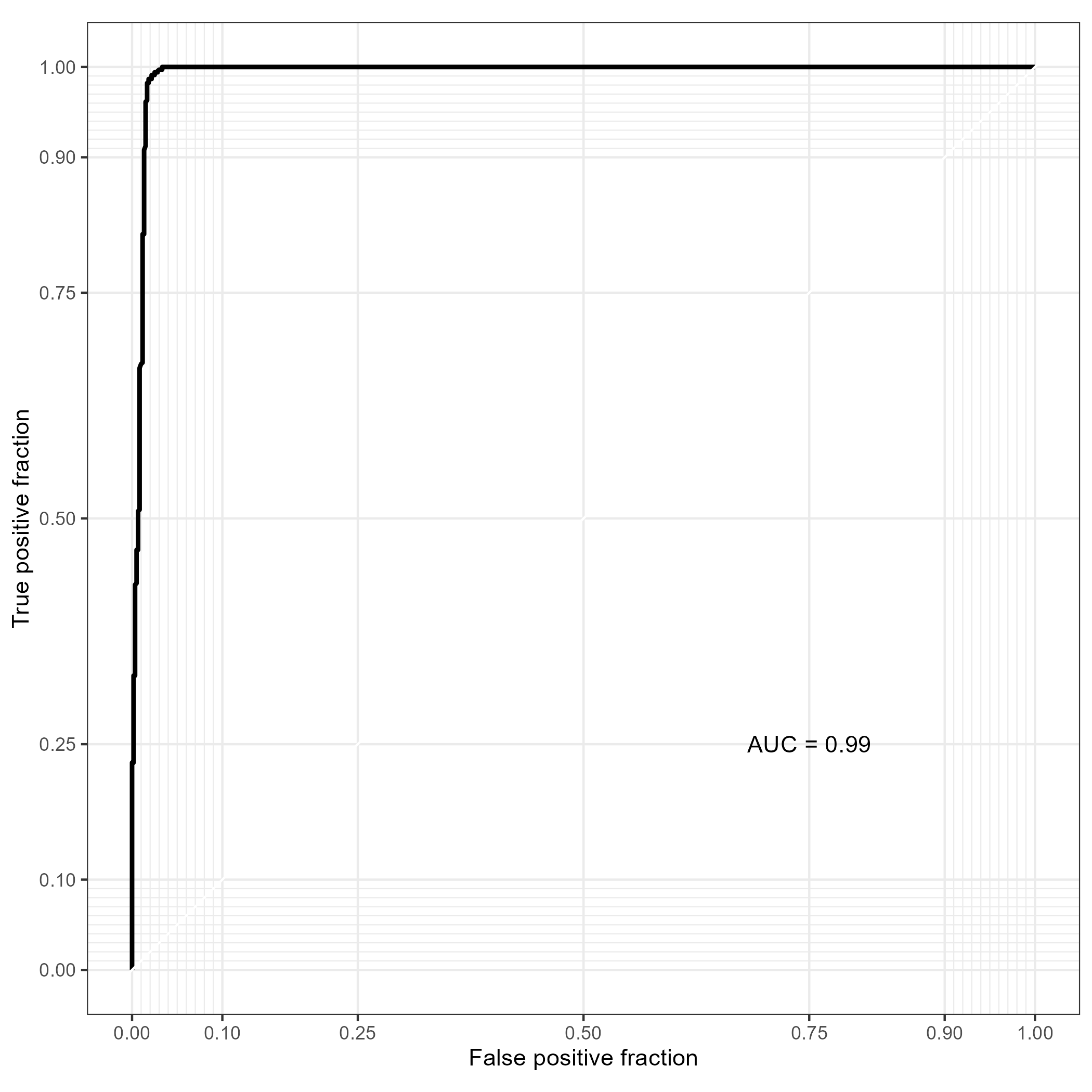
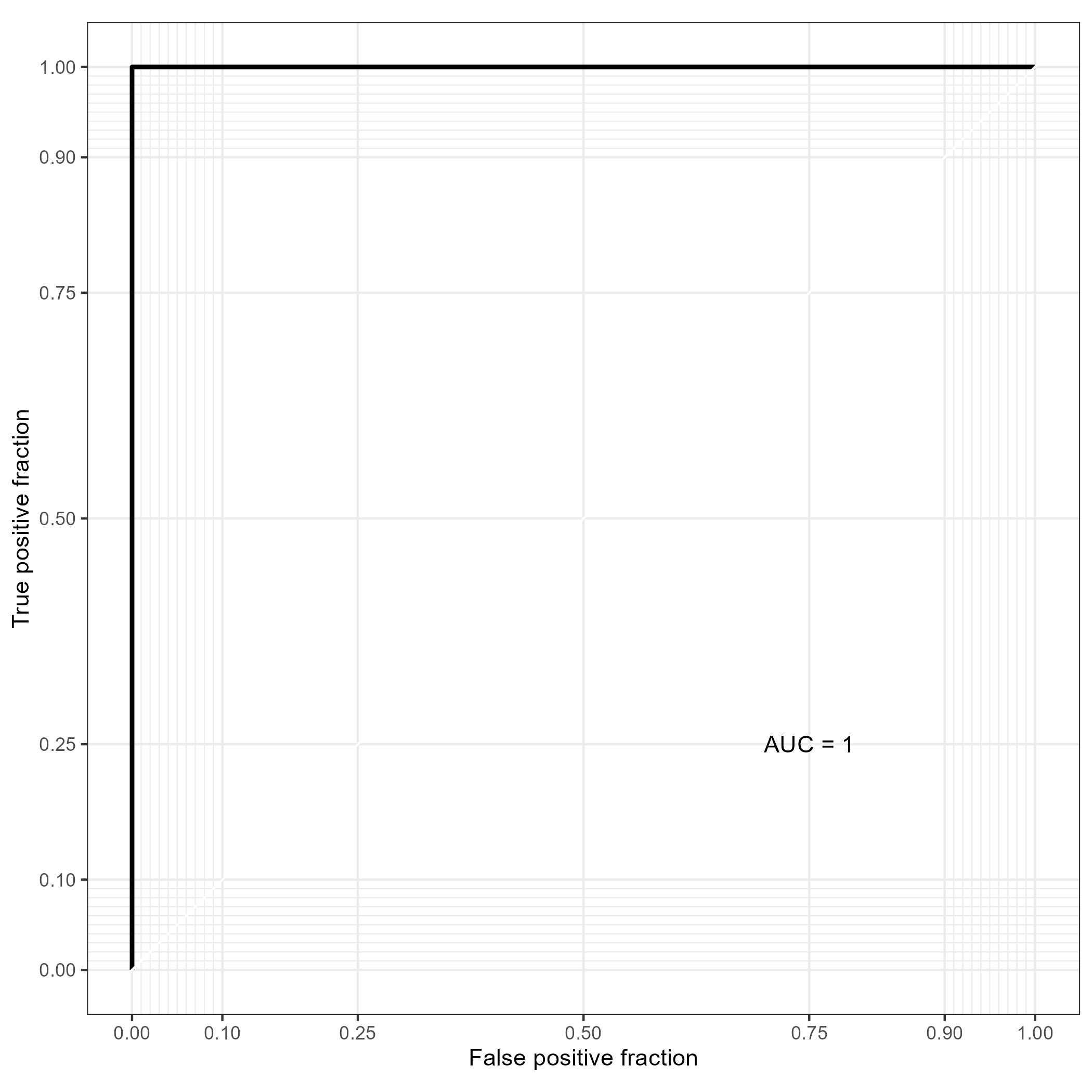
- AUC가 1인 것은 예제 데이터가 검증데이터를 완벽하게 분류한다는 것이다(정확도 100%). 이는 데이터의 수가 적고 매우 구분되는 그룹이기 때문에 이를 기준으로 학습되어 Overfitting 된 것으로 보인다.
- 데이터에 따라 다르지만, 마이크로바이옴 데이터의 경우 논문에서는 적어도 0.8 이상의 AUC가 나와야 한다.
Relative abundance and Genus level
LR.2 <- microbiomeMarker::run_sl(
ps = ps,
norm = "TSS", # total sum scaling = relative abundance
group = "body.site",
method = "LR",
taxa_rank = "Genus")
microbiomeMarker::plot_sl_roc(LR.2, group = "body.site")
ggsave("./Marker_LR02_roc.png")
microbiomeMarker::plot_ef_bar(LR.2)
ggsave("./Marker_LR02_bar.png")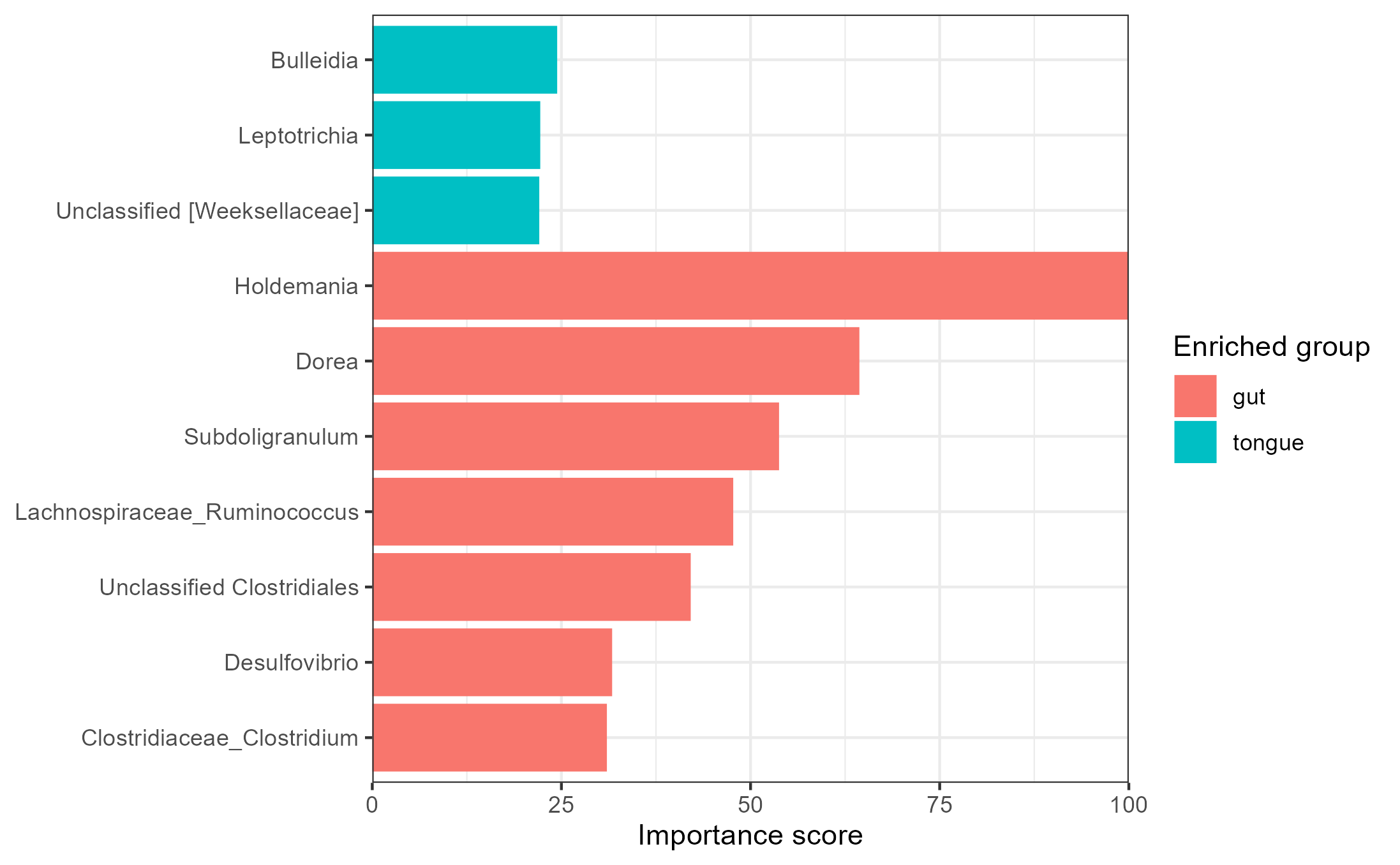
Support vector machine
Species and CLR normalization
SVM <- microbiomeMarker::run_sl(
ps = ps,
norm = "CLR", # central log ratio
group = "body.site",
method = "SVM",
taxa_rank = "Species",
nfolds = 2,
nrepeats = 1,
top_n = 10)
microbiomeMarker::plot_sl_roc(SVM, group = "body.site")
microbiomeMarker::plot_ef_bar(SVM)- nfolds = The number of splits in CV = 교차 검증 시 몇 개의 fold(그룹)으로 나눌지 결정, 2 값이니 전체 데이터를 두 그룹으로 나누어 각 데이터를 검증, 나머지를 학습 데이터로 사용
- nrepeats = the number of complete sets of folds to compute = 교차검증 횟수
=> nfold * nrepeat 해서 총 2회 검증
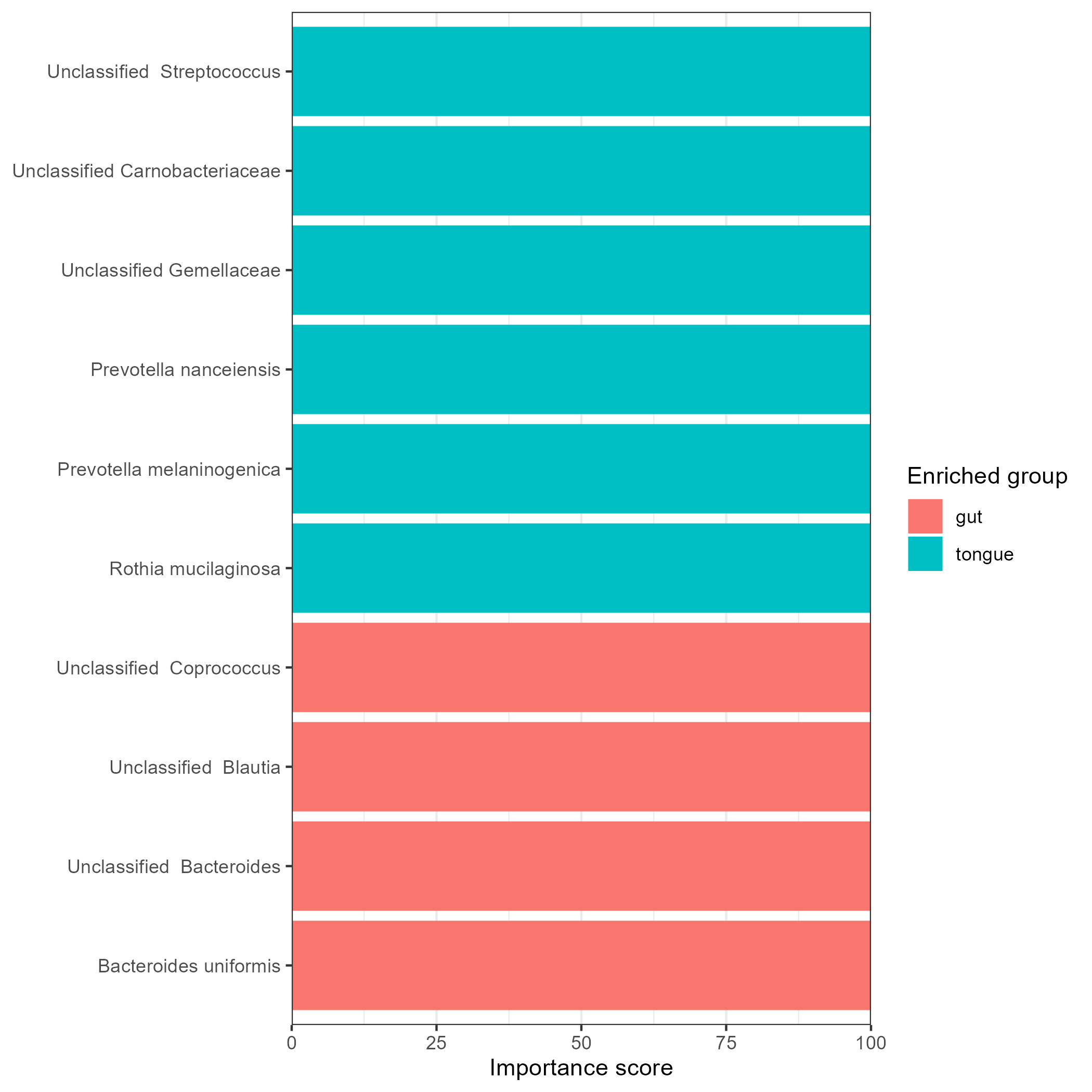
- species가 모두 100의 importance score를 띈 것은 샘플 수가 적고, 매우 구분되는 샘플이기 때문에, vector가 명확하게 샘플들을 구분해서 그런 것 같다. 물론 과적합이라고도 할 수 있다.
Random forest
- 예제 데이터에서는 importance가 계산되지 않음
Rf.1 <- microbiomeMarker::run_sl(
ps = ps,
group = "body.site",
method = "RF")
- 참고로 대부분의 마이크로바이옴 연구에서는 xgboost 같은 앙상블이나 Random forest기법이 쓰인다. LR, SVM은 약간 한물 간 친구로 취급받는다ㅠ
5. 결론
머신러닝, AI, 딥 러닝은 기존의 모든 문제를 해결할 수 있는 key로 취급되어 여러 기사가 쏟아져 나오고 있다. 그러나 이러한 분석은 존재해 왔던 통계 분야의 한 갈래일 뿐이며, 컴퓨터 기술의 발전에 의해 각광받는 분야가 되었다. 본인도 이들을 배우지 않으면 도태된다는 분위기에 휩쓸려 여러 자격증을 공부했다. 그러나 여러 자격증은 실제 분야에서 적용되는 모습과는 거리가 멀었다. 개인적으로 이들을 자격증이라고 부를 수 있는지도 반신반의하다.
서울대 백민경 교수님 말에 따르면, 같은 분야에서 이미 어떻게 활용되고 있는지를 알아보고, 기존에 사용되고 있는 인공지능 모델을 다뤄보는 것으로 공부를 시작하는 것이 가장 좋다고 말씀하였다. 이에 마이크로바이옴 관련 분야에 사용되고 있는 ML 방법을 알아보고 정리해 보았다. 각종 이론에 대해 공부할수록 ML에 사용되는 분석기법들은 기존 마이크로바이옴 분석에 사용되고 있었음을 깨달았다. 활용법을 먼저 알고 역으로 이론을 공부하니 각 분석 방법이 훨씬 잘 와닿았다.
또한 이 글에서는 MicrobiomeMarker 패키지를 통해 간단한 SL을 사용하여 biomarker를 직접 구해보았다.
물론 마이크로바이옴 데이터에 모든 ML 방법을 적용하기엔 한계가 있다. 예를 들어 deep learning의 경우 적어도 만 개 이상의 데이터에 적용해야 한다. 머신러닝은 그보다 적은 데이터를 사용해 결과를 낼 수 있지만, 데이터의 구조에 따라 결과가 상이하다. 또한 분석하고자 하는 데이터의 특성을 잘 알아야 한다. 이를 통해 머신러닝의 방법론만 잘 아는 것이 중요한 게 아니라, 질 좋은 데이터를 구축해야 하며, 데이터 특성을 잘 아는 것이 더 중요하다고 생각된다. 지극히 개인적인 생각이지만, 특정 데이터 분석의 전문가가 더욱 중요해질 것으로 사료된다.
6. 참고
- Marcos-Zambrano, L. J., Karaduzovic-Hadziabdic, K., Loncar Turukalo, T., Przymus, P., Trajkovik, V., Aasmets, O., Berland, M., Gruca, A., Hasic, J., Hron, K., Klammsteiner, T., Kolev, M., Lahti, L., Lopes, M. B., Moreno, V., Naskinova, I., Org, E., Paciência, I., Papoutsoglou, G., Shigdel, R., … Truu, J. (2021). Applications of Machine Learning in Human Microbiome Studies: A Review on Feature Selection, Biomarker Identification, Disease Prediction and Treatment. Frontiers in microbiology, 12, 634511. https://doi.org/10.3389/fmicb.2021.634511
- Hernández Medina, R., Kutuzova, S., Nielsen, K.N. et al. Machine learning and deep learning applications in microbiome research. ISME COMMUN. 2, 98 (2022). https://doi.org/10.1038/s43705-022-00182-9
- 또 다른 microbiome 데이터에 ML을 적용하는 패키지 : https://cran.r-project.org/web/packages/mikropml/vignettes/introduction.html ( from mothur 개발 연구실)
- 패키지를 사용하지 않고 random forest분석하기 : https://rpubs.com/michberr/randomforestmicrobe
- 사람 장 마이크로바이옴 데이터를 Random Forest와 XGBoost를 통해 예측 코드 : https://www.kaggle.com/code/antaresnyc/prediction-with-random-forest-and-xgboost
영어를 배우려면 관심 있는 콘텐츠를 보라는 말처럼, AI나 ML을 배우려면 본인이 관심이 있는 데이터를 분석해 보는 게 좋을 것 같다. 만약 농구에 관심이 있다면 MBA선수들의 신장, 키, 몸무게에 대한 승률분석이라던가. 각 선수들의 이적료에 대한 예측 같은 것 말이다.
물론 현재 AI분야의 기록은 놀라울 정도이다. 특히 창조적인 분야(이미지, 글 생산)나, 차량용 영상인식기술, 암의 여러 지표를 예측하는 의료 빅데이터 분석, 유전체 바이오마커를 검출하는 딥러닝 모델(e.g.Hilbert-CNN )등은 여기에 사용되는 방식은 이 글에 적은 것보다 훨씬 더 복잡하다. 우리는 단순히 이 단계까지 갈 순 없겠지만, 차근차근 공부해 가면 이해는 할 수 있지 않을까 생각해 본다.