
작성 : 2023.06.21
| 목표
마이크로바이옴 데이터로 여러 종류의 상관관계 그림을 그려보자. 특히 Heatmap에 집중해서 관찰하자!
| 예제 데이터
- qiime2 moving pictures Tutorial에 나오는 데이터로, 사람의 4 부위에 해당하는 마이크로바이옴 데이터를 담고 있다.
- 이 데이터는 phyloseq 데이터로, biom형식으로 구성되어 있다. [참고]
| 시각화의 종류
크게 Scatter plot과 Heatmap으로 나눌 수 있다.
Scatter plot은 두 변수 간의 관계를 나타내고 싶을 때 사용한다. 이에 관한 글은 Regression plot에 정리해 두었다.
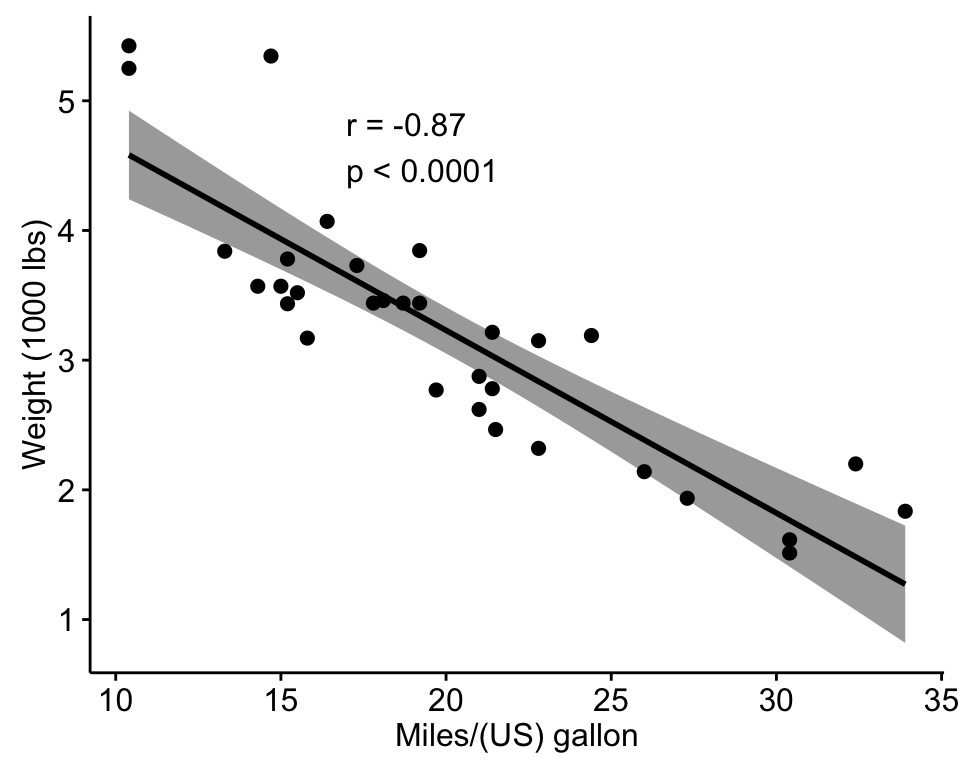
Heatmap은 여러 변수 간의 상관관계를 보고 싶을 때 사용한다.
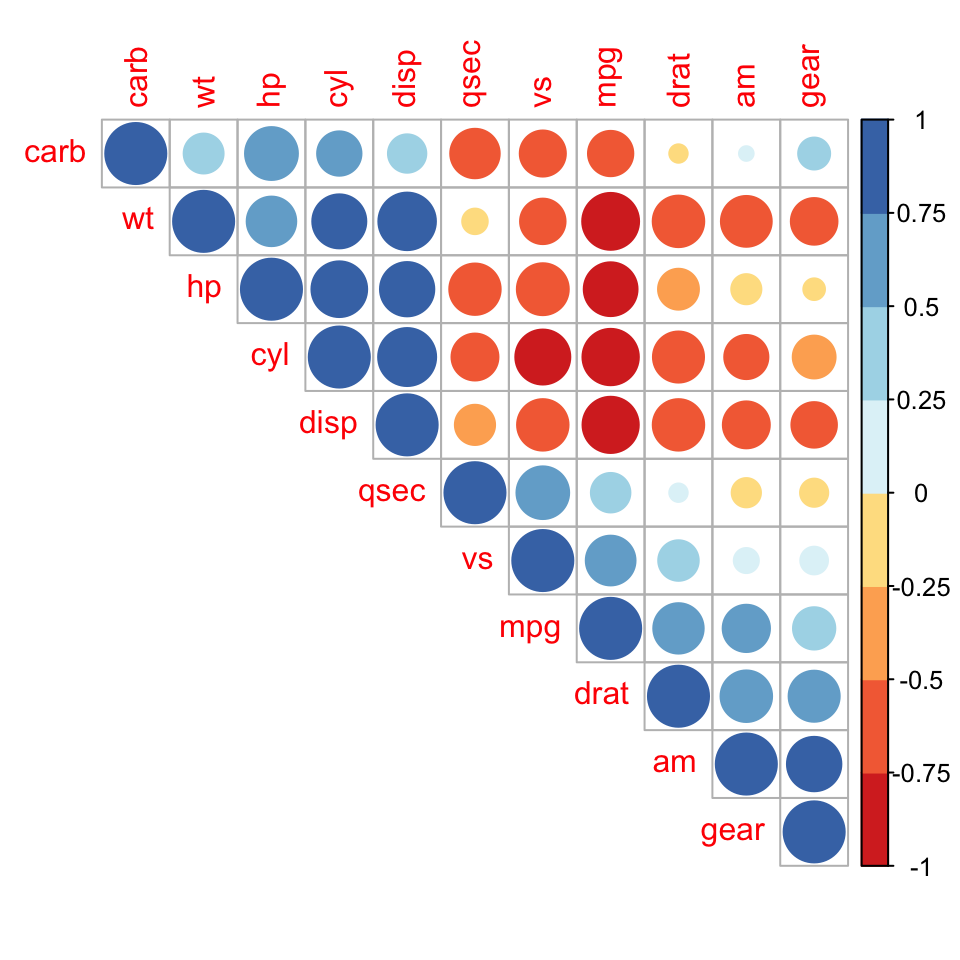
| 시각화
0. 데이터 전처리
library(phyloseq)
library(ggplot2)
library(ggpubr)
library(ggrepel)
library(tibble)
library(dplyr)
ps <- readRDS("./ps.rds")
# phyloseq-class experiment-level object
# otu_table() OTU Table: [ 770 taxa and 34 samples ]
# sample_data() Sample Data: [ 34 samples by 8 sample variables ]
# tax_table() Taxonomy Table: [ 770 taxa by 7 taxonomic ranks ]
# phy_tree() Phylogenetic Tree: [ 770 tips and 768 internal nodes ]
ps.rel <- transform_sample_counts(ps, function(x) x/sum(x) )
Alpha diversity계산하기
alpha_div <- estimate_richness(ps.rel, measures = "Shannon")
alpha_meta <- merge(sample_data(ps.rel), alpha_div, by = "row.names")
1. Scatter plot에 p-value와 상관계수 표시하기
- 기존의 Regression plot에서 회귀분석이 아닌 상관분석을 시행해 봅시다.
- 예제데이터에서 Alpha diversity와 "days.since.experiment.start"변수간의 상관관계를 보자. (큰 의미는 없다.)
p <- ggplot(alpha_meta, aes(days.since.experiment.start, Shannon, label = Row.names)) +
geom_point(aes(shape = body.site, color = body.site), size = 3) +
stat_smooth(color = "blue", method = "lm") +
ggpubr::stat_cor(method = "spearman", label.x = 1, label.y = 4.5, color = "red") +
theme_test() +
labs(y = "Shannon", x = "Day") +
geom_text_repel(size = 3)
p
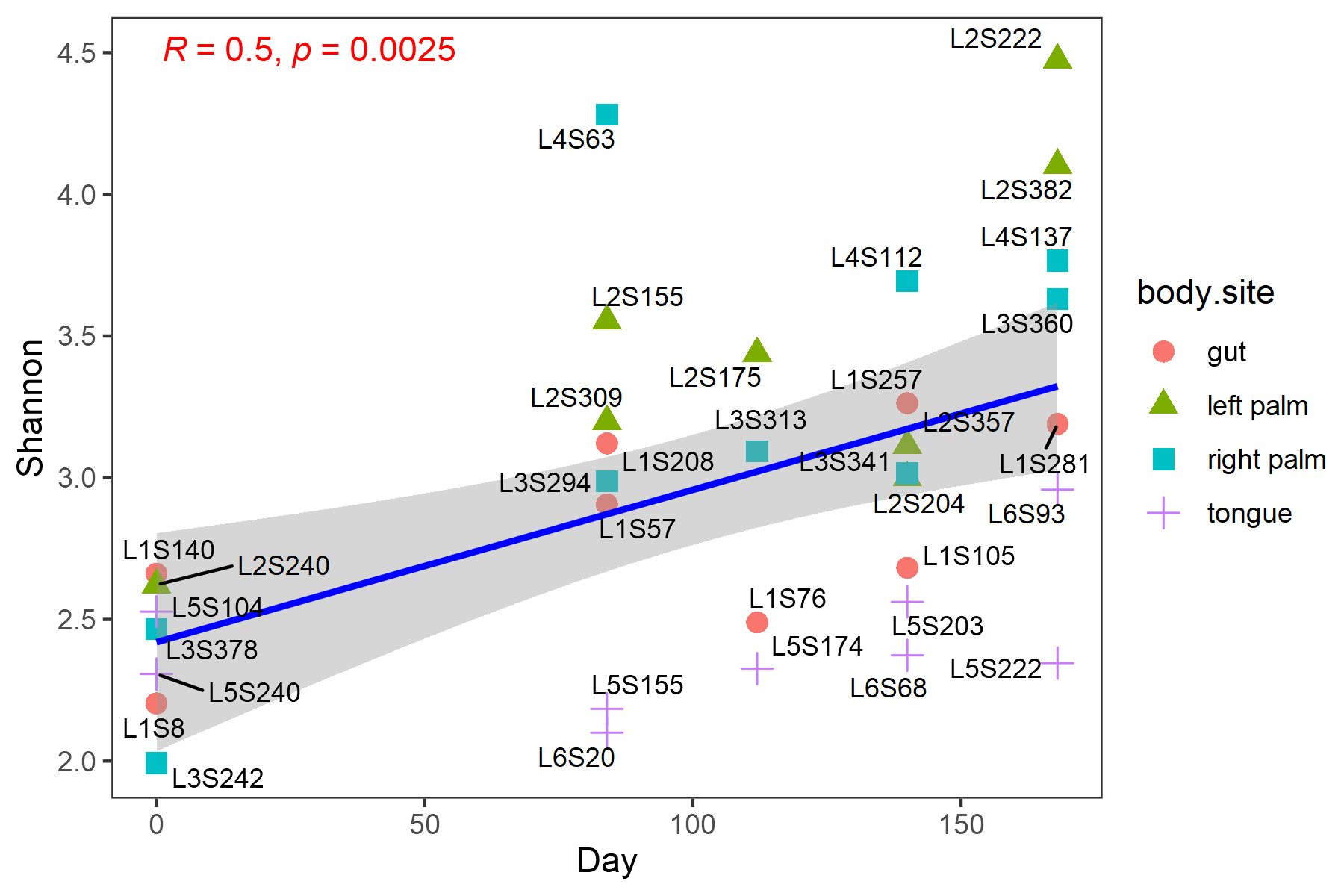
날이 지날수록 샘플의 shannon 다양성은 증가하였으며, 상관계수는 0.5 값을 가지고 p-value값은 0.0025 값을 가집니다.
+) 그림에 p-value값을 표현하는 것으로 (1) 수동으로 x, y축을 정해서 삽입하는 방법과 (2) ggpurb를 이용하거나 (3) ggpmisc 패키지를 사용하는 방법이 대표적이다. 그중에서 제일 간단한 ggpurb 패키지를 추천합니다.
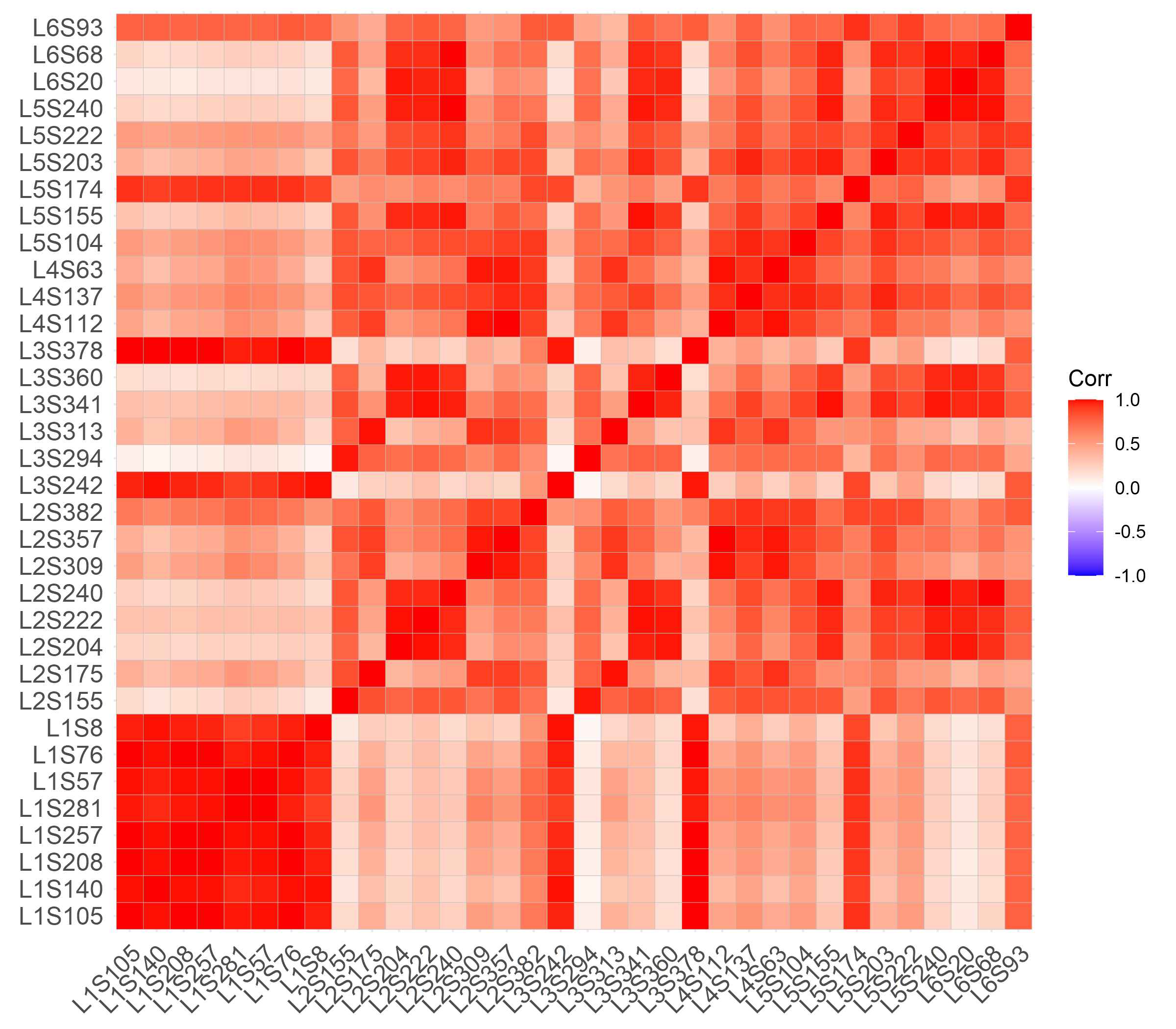
2. Heatmap으로 각 샘플 간의 상관관계 보기
어떤 패키지를 사용하는지 중요하진 않지만, 이번에는 ggcorrplot을 사용해 보겠습니다.
먼저, 데이터를 사용하기 편하게 만들어 줍니다.
library(reshape2)
ps.ph <- tax_glom(ps.rel, taxrank = "Phylum")
TAX <- tax_table(ps.ph) %>% data.frame()
OTU <- otu_table(ps.ph) %>% data.frame()
META <- sample_data(ps.ph) %>% data.frame()
Table <- merge(TAX, OTU, by = "row.names")
Table
ggcorrplot::ggcorrplot(cor(Table[, 9:42]))
3. Heatmap으로 각 샘플이 속해있는 그룹에서 비교하기
예제 데이터는 gut, tongue과 right, left palm의 데이터를 담고 있습니다.
이 그룹 간의 비교를 하려고 합니다. 이때 각 그룹이 속해있는 샘플의 Relative abundance를 각 샘플링 부위 별로 합해주어야 합니다.
OTU.t <- otu_table(ps.ph) %>% t() %>% as.matrix() %>% data.frame()
Table2 <- merge(META[,c("body.site"), drop = F], OTU.t, by = "row.names")
Table2.Site <- Table2[, -1] %>% group_by(body.site) %>%
summarise_each(funs(sum)) %>% tibble::column_to_rownames("body.site") %>% t()
ggcorrplot::ggcorrplot(cor(Table2.Site))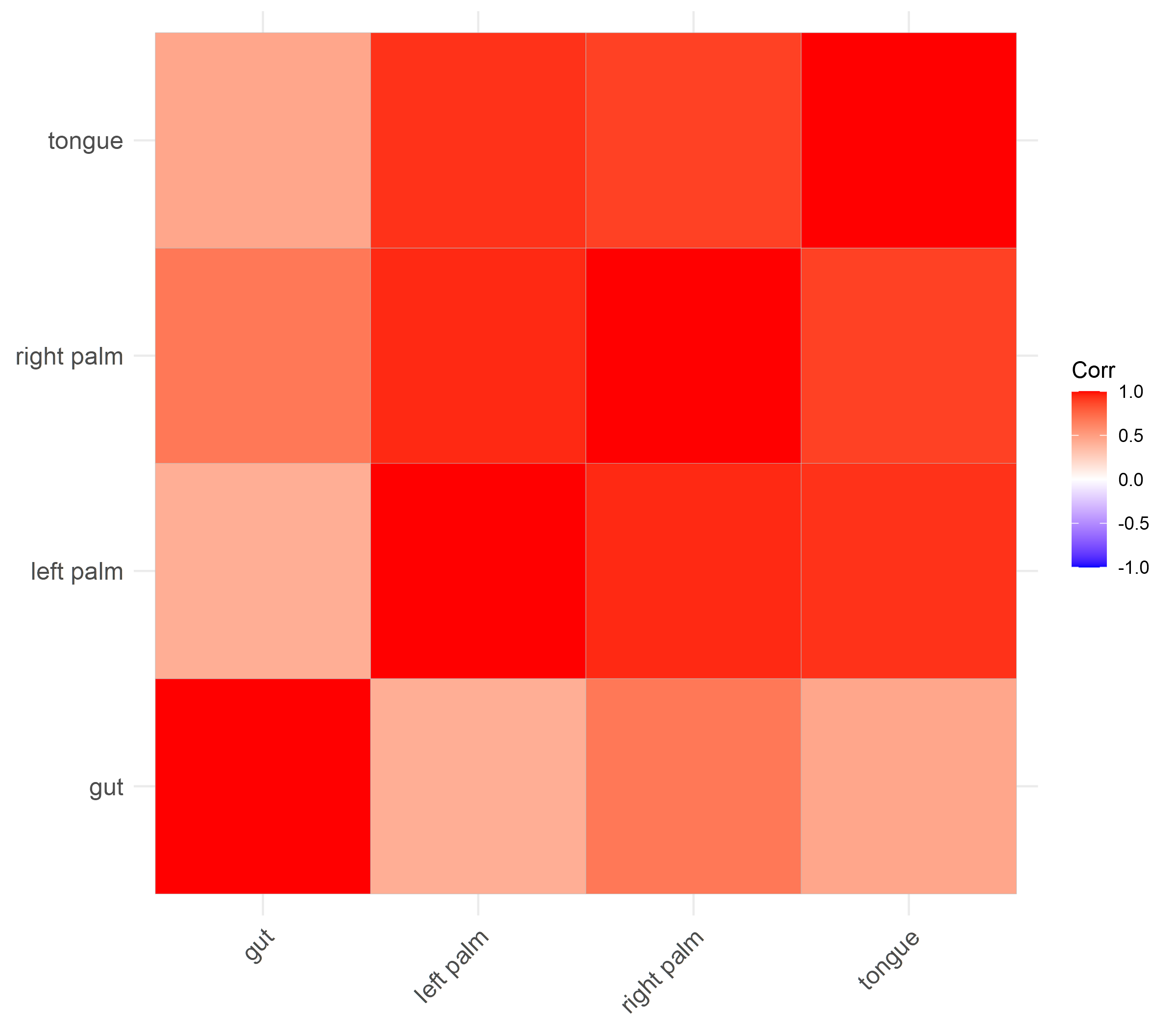
4. Heatmap으로 각 Phylum 간의 상관관계 보기
Table.3 <- Table[, c(3, 9:42)] %>% column_to_rownames("Phylum") %>% t()
ggcorrplot::ggcorrplot(cor(Table.3))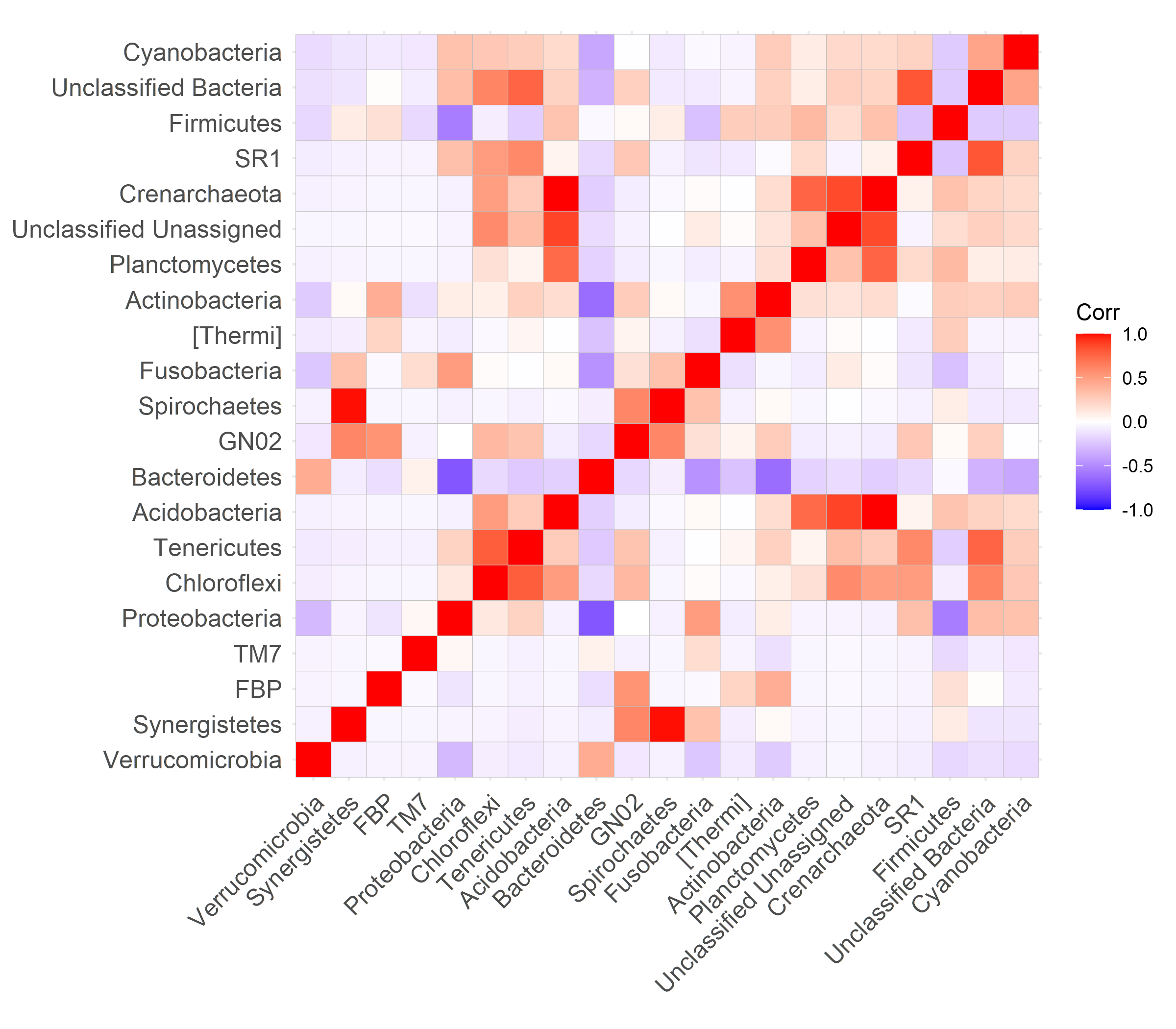
4. Heatmap으로 Metadata와 Phylum 간의 상관관계 보기
OTU.t <- otu_table(ps.ph) %>% t() %>% as.matrix() %>% data.frame()
Table.4 <- merge(META[,c("days.since.experiment.start"), drop = F],
OTU.t, by = "row.names") %>%
tibble::column_to_rownames("Row.names")
Cor.4 <- cor(Table.4)["days.since.experiment.start",colnames(OTU.t), drop = F]
ggcorrplot::ggcorrplot(Cor.4)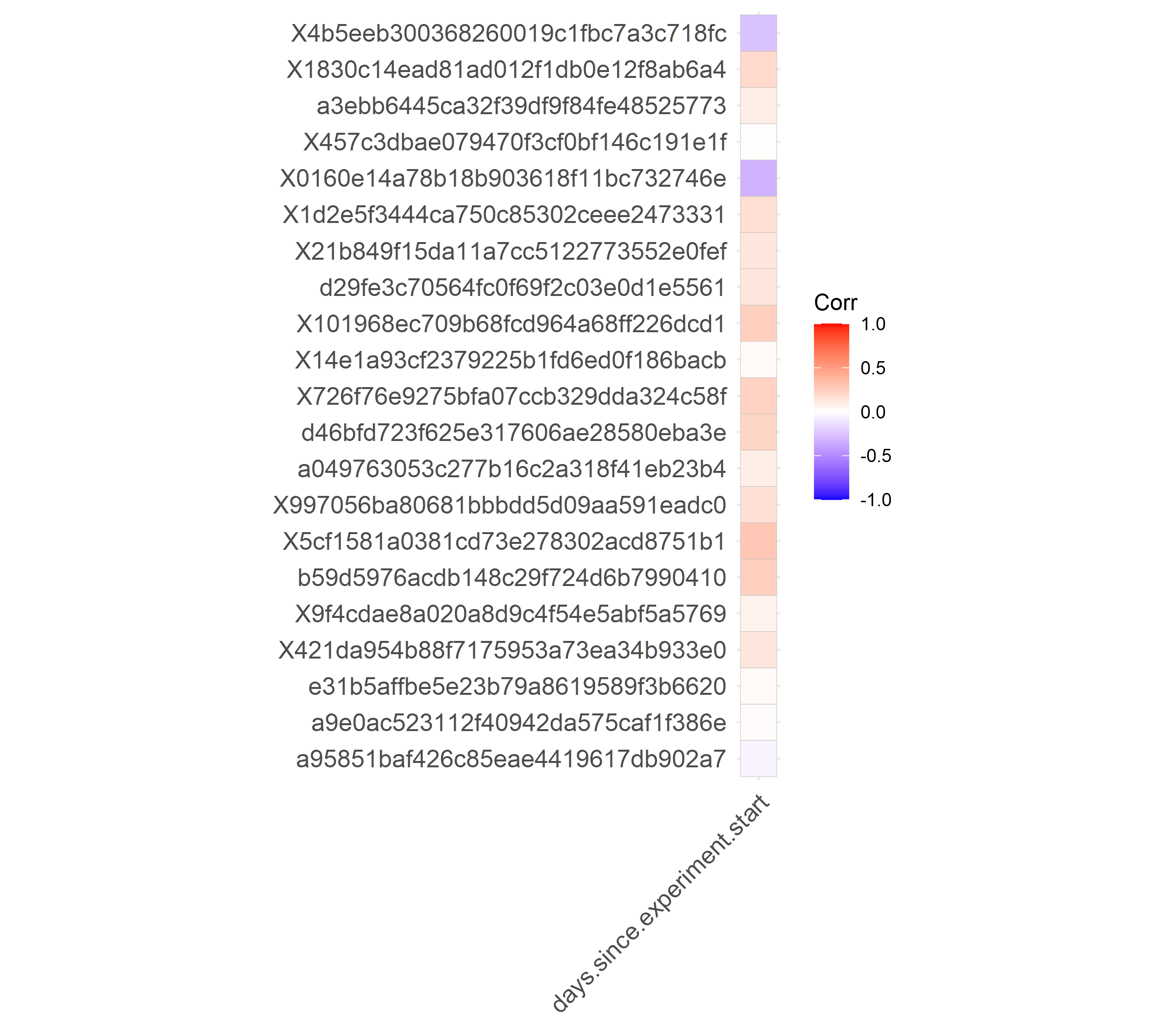
t() 과정에서 숫자로 시작하는 OTU ID에 "X"문자가 추가된다. 이는 간단하게 아래 명령어로 해결 가능하다
names(table) <- sub("^X", "", names(table))+) 기타 방법
5. Heatmap으로 PICRUSt2 결과와 Phylum 간의 상관관계 보기
아래 예제 파일을 사용해서 Picrust2 결과와 Phylum 간의 상관관계 그래프를 그려보자
library(readr)
kegg_abundance.30 <- read_csv("./Kegg_example.csv") %>%
column_to_rownames("...1")
TAX.ph <- as.data.frame(tax_table(tax_glom(ps.rel, taxrank = "Phylum")))
OTU.ph <- as.data.frame(otu_table(tax_glom(ps.rel, taxrank = "Phylum")))
table.ph <- merge(TAX.ph, OTU.ph, by = "row.names")
table.ph.2 <- table.ph[ ,c(3, 9:42)] %>% column_to_rownames("Phylum") %>% t()
Table.final <- merge(table.ph.2, kegg_abundance.30, by = "row.names") %>% column_to_rownames("Row.names")
Cor <- cor(Table.final)
Cor.2 <- Cor[colnames(table.ph.2), colnames(kegg_abundance.30)]
ggcorrplot::ggcorrplot(Cor.2)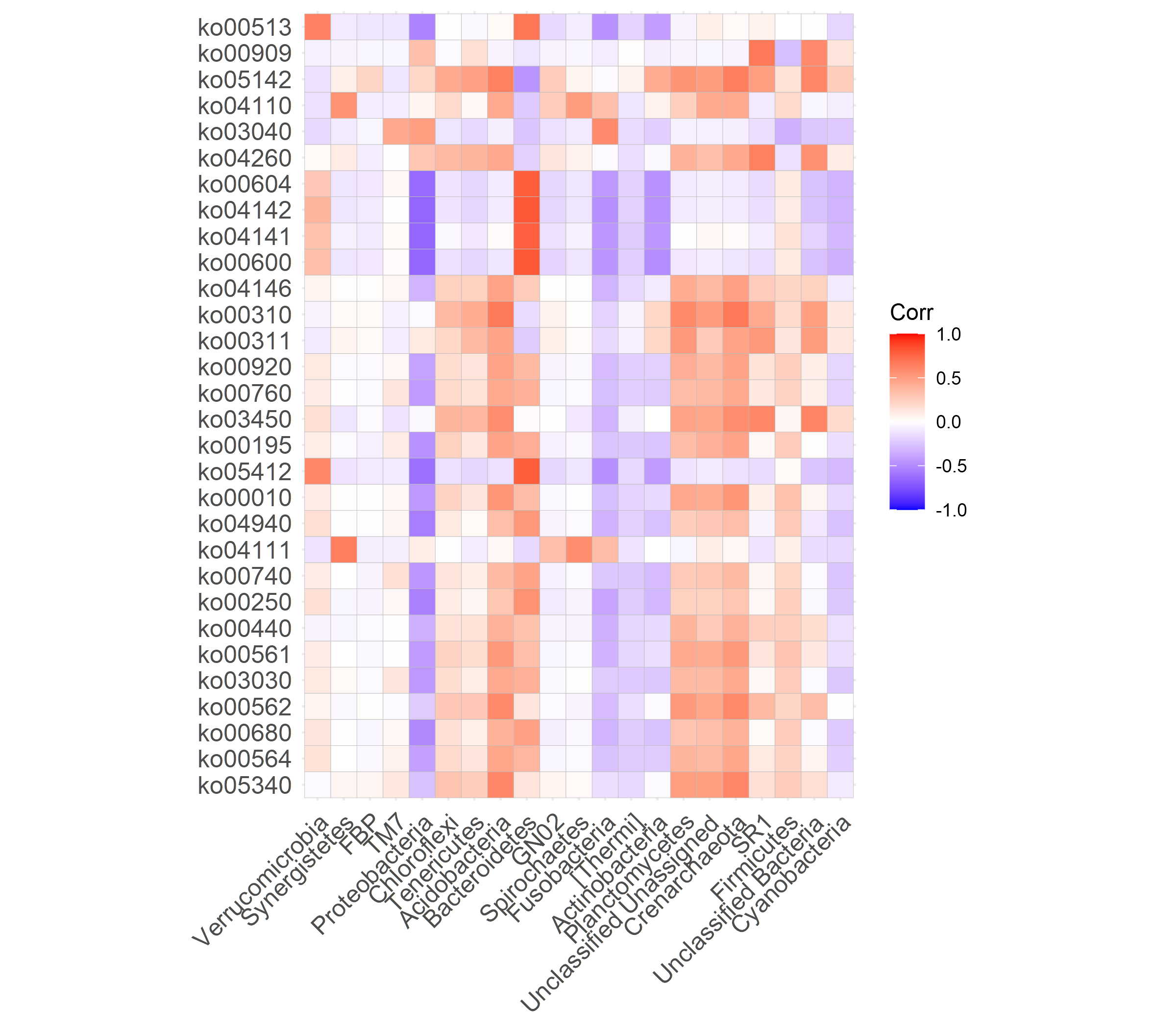
+) Heatmap에 통계적 유의성 표시하는 방법
ggcorrplot에서는 통계적 유의성을 *이 아닌 X로 표현한다.
p_values <- cor_pmat(Cor)[colnames(table.ph.2), colnames(kegg_abundance.30)]
dim(p_values)
p <- ggcorrplot::ggcorrplot(Cor.2,
method = c("circle"),
# type = c("lower"),
p.mat= p_values,
insig = "pch"
)
p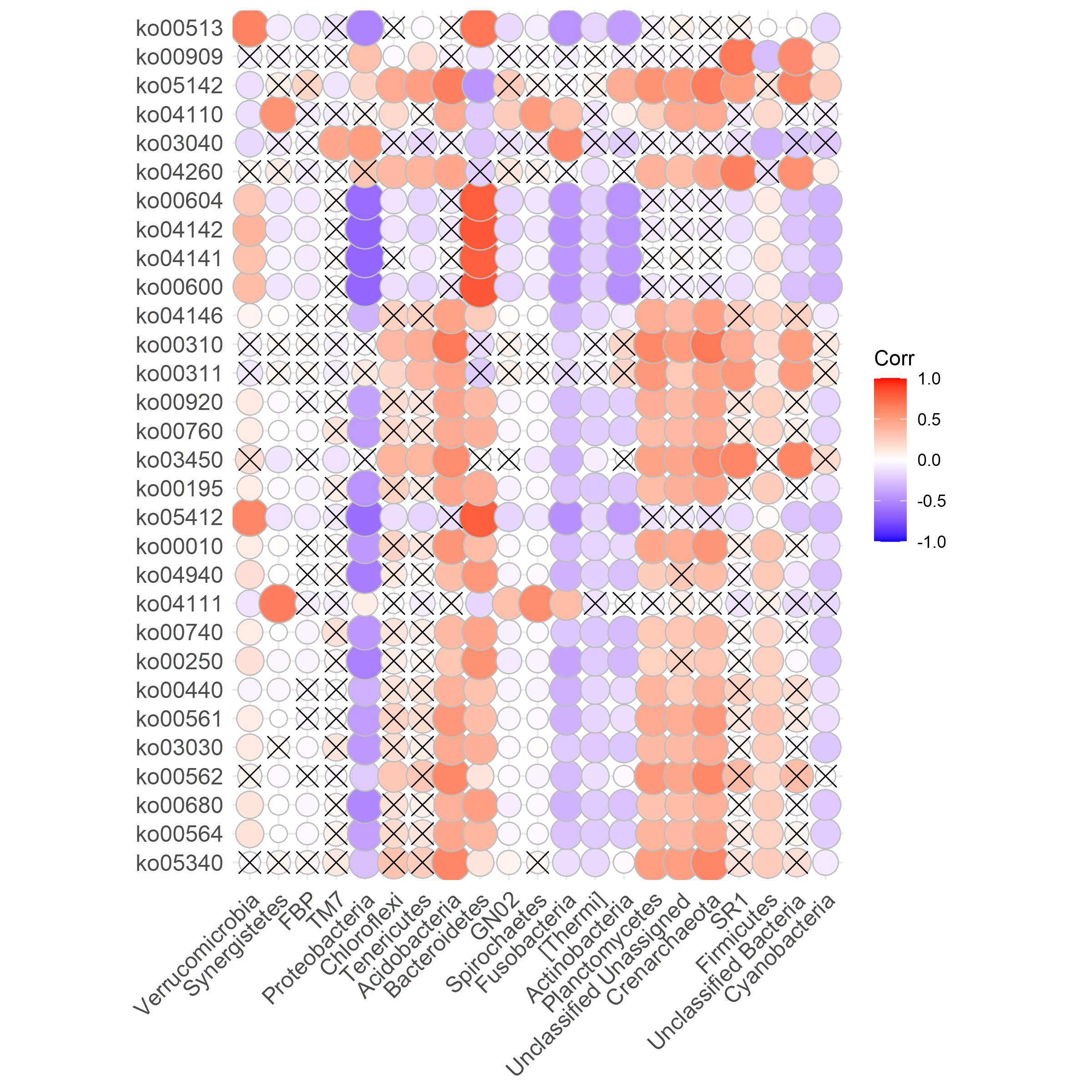
| 참고
http://www.sthda.com/english/wiki/correlation-analyses-in-r
작성 : 2023.06.21
| 목표
마이크로바이옴 데이터로 여러 종류의 상관관계 그림을 그려보자. 특히 Heatmap에 집중해서 관찰하자!
| 예제 데이터
- qiime2 moving pictures Tutorial에 나오는 데이터로, 사람의 4 부위에 해당하는 마이크로바이옴 데이터를 담고 있다.
- 이 데이터는 phyloseq 데이터로, biom형식으로 구성되어 있다. [참고]
| 시각화의 종류
크게 Scatter plot과 Heatmap으로 나눌 수 있다.
Scatter plot은 두 변수 간의 관계를 나타내고 싶을 때 사용한다. 이에 관한 글은 Regression plot에 정리해 두었다.
Heatmap은 여러 변수 간의 상관관계를 보고 싶을 때 사용한다.
| 시각화
0. 데이터 전처리
library(phyloseq)
library(ggplot2)
library(ggpubr)
library(ggrepel)
library(tibble)
library(dplyr)
ps <- readRDS("./ps.rds")
# phyloseq-class experiment-level object
# otu_table() OTU Table: [ 770 taxa and 34 samples ]
# sample_data() Sample Data: [ 34 samples by 8 sample variables ]
# tax_table() Taxonomy Table: [ 770 taxa by 7 taxonomic ranks ]
# phy_tree() Phylogenetic Tree: [ 770 tips and 768 internal nodes ]
ps.rel <- transform_sample_counts(ps, function(x) x/sum(x) )
Alpha diversity계산하기
alpha_div <- estimate_richness(ps.rel, measures = "Shannon")
alpha_meta <- merge(sample_data(ps.rel), alpha_div, by = "row.names")
1. Scatter plot에 p-value와 상관계수 표시하기
- 기존의 Regression plot에서 회귀분석이 아닌 상관분석을 시행해 봅시다.
- 예제데이터에서 Alpha diversity와 "days.since.experiment.start"변수간의 상관관계를 보자. (큰 의미는 없다.)
p <- ggplot(alpha_meta, aes(days.since.experiment.start, Shannon, label = Row.names)) +
geom_point(aes(shape = body.site, color = body.site), size = 3) +
stat_smooth(color = "blue", method = "lm") +
ggpubr::stat_cor(method = "spearman", label.x = 1, label.y = 4.5, color = "red") +
theme_test() +
labs(y = "Shannon", x = "Day") +
geom_text_repel(size = 3)
p
날이 지날수록 샘플의 shannon 다양성은 증가하였으며, 상관계수는 0.5 값을 가지고 p-value값은 0.0025 값을 가집니다.
+) 그림에 p-value값을 표현하는 것으로 (1) 수동으로 x, y축을 정해서 삽입하는 방법과 (2) ggpurb를 이용하거나 (3) ggpmisc 패키지를 사용하는 방법이 대표적이다. 그중에서 제일 간단한 ggpurb 패키지를 추천합니다.
2. Heatmap으로 각 샘플 간의 상관관계 보기
어떤 패키지를 사용하는지 중요하진 않지만, 이번에는 ggcorrplot을 사용해 보겠습니다.
먼저, 데이터를 사용하기 편하게 만들어 줍니다.
library(reshape2)
ps.ph <- tax_glom(ps.rel, taxrank = "Phylum")
TAX <- tax_table(ps.ph) %>% data.frame()
OTU <- otu_table(ps.ph) %>% data.frame()
META <- sample_data(ps.ph) %>% data.frame()
Table <- merge(TAX, OTU, by = "row.names")
Table
ggcorrplot::ggcorrplot(cor(Table[, 9:42]))
3. Heatmap으로 각 샘플이 속해있는 그룹에서 비교하기
예제 데이터는 gut, tongue과 right, left palm의 데이터를 담고 있습니다.
이 그룹 간의 비교를 하려고 합니다. 이때 각 그룹이 속해있는 샘플의 Relative abundance를 각 샘플링 부위 별로 합해주어야 합니다.
OTU.t <- otu_table(ps.ph) %>% t() %>% as.matrix() %>% data.frame()
Table2 <- merge(META[,c("body.site"), drop = F], OTU.t, by = "row.names")
Table2.Site <- Table2[, -1] %>% group_by(body.site) %>%
summarise_each(funs(sum)) %>% tibble::column_to_rownames("body.site") %>% t()
ggcorrplot::ggcorrplot(cor(Table2.Site))
4. Heatmap으로 각 Phylum 간의 상관관계 보기
Table.3 <- Table[, c(3, 9:42)] %>% column_to_rownames("Phylum") %>% t()
ggcorrplot::ggcorrplot(cor(Table.3))
4. Heatmap으로 Metadata와 Phylum 간의 상관관계 보기
OTU.t <- otu_table(ps.ph) %>% t() %>% as.matrix() %>% data.frame()
Table.4 <- merge(META[,c("days.since.experiment.start"), drop = F],
OTU.t, by = "row.names") %>%
tibble::column_to_rownames("Row.names")
Cor.4 <- cor(Table.4)["days.since.experiment.start",colnames(OTU.t), drop = F]
ggcorrplot::ggcorrplot(Cor.4)t() 과정에서 숫자로 시작하는 OTU ID에 "X"문자가 추가된다. 이는 간단하게 아래 명령어로 해결 가능하다
names(table) <- sub("^X", "", names(table))+) 기타 방법
5. Heatmap으로 PICRUSt2 결과와 Phylum 간의 상관관계 보기
아래 예제 파일을 사용해서 Picrust2 결과와 Phylum 간의 상관관계 그래프를 그려보자
library(readr)
kegg_abundance.30 <- read_csv("./Kegg_example.csv") %>%
column_to_rownames("...1")
TAX.ph <- as.data.frame(tax_table(tax_glom(ps.rel, taxrank = "Phylum")))
OTU.ph <- as.data.frame(otu_table(tax_glom(ps.rel, taxrank = "Phylum")))
table.ph <- merge(TAX.ph, OTU.ph, by = "row.names")
table.ph.2 <- table.ph[ ,c(3, 9:42)] %>% column_to_rownames("Phylum") %>% t()
Table.final <- merge(table.ph.2, kegg_abundance.30, by = "row.names") %>% column_to_rownames("Row.names")
Cor <- cor(Table.final)
Cor.2 <- Cor[colnames(table.ph.2), colnames(kegg_abundance.30)]
ggcorrplot::ggcorrplot(Cor.2)
+) Heatmap에 통계적 유의성 표시하는 방법
ggcorrplot에서는 통계적 유의성을 *이 아닌 X로 표현한다.
p_values <- cor_pmat(Cor)[colnames(table.ph.2), colnames(kegg_abundance.30)]
dim(p_values)
p <- ggcorrplot::ggcorrplot(Cor.2,
method = c("circle"),
# type = c("lower"),
p.mat= p_values,
insig = "pch"
)
p
| 참고
http://www.sthda.com/english/wiki/correlation-analyses-in-r