
글 작성 (2024-05-24 ~ 2024-05-28)
좀 더 읽기 쉬운 글로 수정하였습니다 (2024-11-19)

모든 생물학도가 사용한다고 해도 과언이 아닌 BLAST, 잘 알고 계시나요?
생물정보학에서 서열정렬(Sequence Alignment)은 필수적인 기술로, 유전학, 단백질 연구, 진화생물학 등 다양한 분야에서 사용되고 있습니다. 이 글에서는 쉽게 이해할 수 있도록 서열 정렬의 역사, 기본 개념, 그리고 대표적인 도구인 BLAST까지 알아볼까요?
1. 서열 정렬(Sequence alignment) 이란?
서열 정렬은 DNA, RNA 또는 단백질의 서열을 배열하여 서열 간의 기능적, 구조적 또는 진화적 연관성을 모두 파악하여 유사성을 확인하는 것이다. 또한 유사성을 통해 우리가 알아보고자 하는 것은 다음과 같습니다.
- 두 유전자의 서열이 얼마나 비슷한가?
- 특정 단백질 서열이 어떤 생물 종에 보존되어 있는가?
- 서열 간의 진화적 관계를 무엇인가?
이를 알아보기 위해 전장 유전체를 정렬할 수도 있고, 마이크로바이옴 연구의 경우 계통 분류를 위한 일부 메타바코딩(metabarcoding) 서열을 기준 균주(type strain)와 비교해 계통을 판별하기도 합니다.
일반적인 정렬방식은 아래와 같이 나눌 수 있습니다.
- Pairwise Alignment vs. Multiple Sequence Alignment
- Global alignment vs. Local alignment
2) Pairwise Alignment와 Multiple Sequence Alignment
- 다중서열정렬(Multiple Sequence Alignment; MSA): 여러 서열 간의 비교 정렬 방식으로, 보존된 영역(conserved regions)을 찾고, 이는 계통학적 관계 추론(계통수 제작) 등에 사용됩니다.
- 대부분 점진적 방법(progressive alignment)나 반복적 방법(iterative refinment)을 사용합니다
- 대표적인 도구로는 ClustalW, MUSCLE 등이 있습니다.
- 쌍서열정렬(Pairwise Alignment): 두 서열 간의 유사성을 평가하고 보존된 영역을 분석하는 정렬방식입니다.
- 크게 글로벌 정렬( Global alignment)과 로컬 정렬(Local alignment) 방식으로 나뉩니다.
- 세부적인 정렬 방식은 크게 Dot-Matrix Methods, Dynamic Programming, Word Methods, dynamic programming, heuristic algorithms, probabilistic methods 등이 있습니다. 또한 이중 일부는 MSA로 확장하여 사용 가능합니다.
- 우리가 알아보고자 하는 BLAST는 word methods 방법을 사용합니다.
1) Global alignment과 Local alignment
- 글로벌 정렬(Global alignment): 서열 비교 시 각 서열의 모든 길이를 비교하여 전체적인 유사도(similarity)를 파악하는 방법입니다. 같은 길이의 서열을 비교할 때 유용합니다.
- e.g. f Needleman and Wunsch
- 로컬 정렬(Local alignment): 가장 유사한 서열이 많은 부위만 비교하는 방법으로, 검색하고자 하는 서열이 데이터베이스에 비해 query 서열이 짧을 때 유용합니다.
- e.g. Smith and Waterman algorithm/ FASTA/ BLAST

2. 서열 정렬의 역사
1) Needleman-Wunsch의 global alignment algorithm (1970)
- 글로벌 정렬을 통해 서열 정렬이 수학적으로 체계화되었습니다. 그러나 전체적인 유사도보다는 보다는 지역적인 유사도가 보다 생물학적인 의미를 지니고 있기 때문에 local alignment 도구의 필요성이 대두되었습니다.
2) Smith-Waterman의 local alignment algorithm (1981)
- dynamic programming 사용하는 알고리즘으로, 짧은 서열 간의 정렬을 정확하게 수행할 수 있게 되었습니다.
- 최적의 비용을 가지는 정렬을 찾아주지만 계산 시간이 오래 걸립니다.
3) FASTA(1988)와 BLAST(1990)
- 이 또한 모든 서열을 조사하는 것이 아닌, 일부 word 중심으로 정렬하는 기법인 word based and heuristic algorithhms를 사용합니다. 이는 아래 BLSAT에서 자세하게 확인해 보겠습니다.
3. 치환 행렬(Substitution matrix)
1) 치환 행렬(Substitution matrix)이란?
뉴클레오타이드의 정렬에는 일치(match)와 불일치(mismatch)를 단순하게 계산합니다. 실제로 BLAST는 match에서 +2, mismatch에서 -3의 점수를 부여하고 있습니다. 그러나 아미노산의 서열 정렬에서는 물리, 화학적 특성을 고려한 치환행렬을 사용해야 합니다.
예시를 통해서 자세히 알아봅시다.
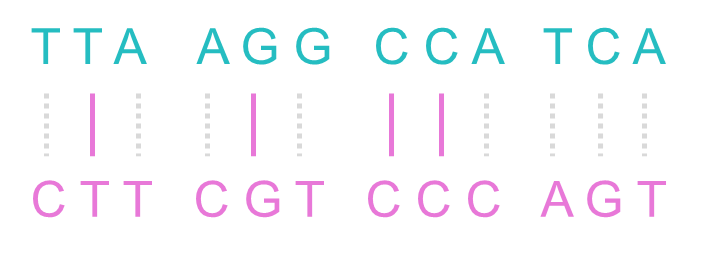
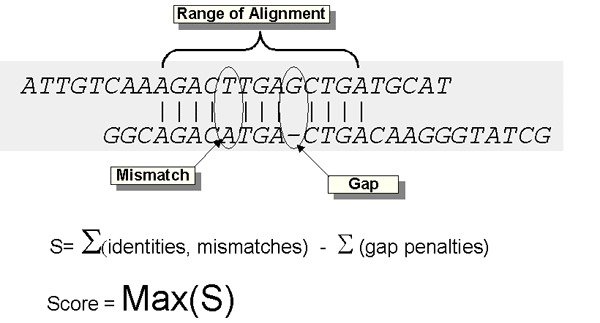
두 서열을 정렬하였을 때, 일치하지 않는 부분을 확인할 수 있습니다.
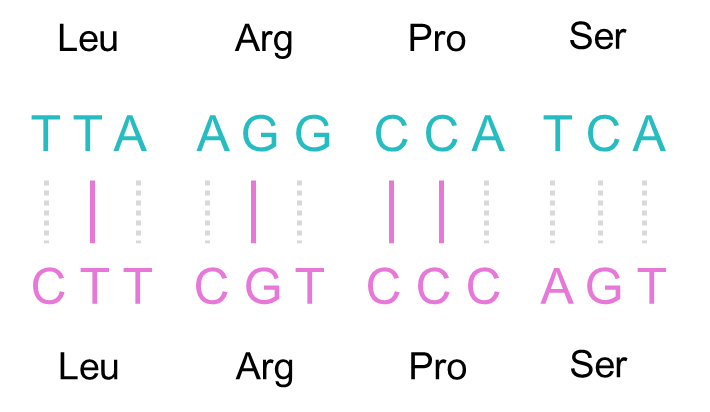
그러나 위, 아래 서열 모두 같은 단백질을 암호화하고 있습니다.
우리는 이때 단순한 서열의 유사성이 아닌 단백질로 번역된 후를 고려해야 합니다.
또한 우리는 단백질의 구조를 고려해야 합니다. 아래의 세 가지 아미노산을 관찰해 봅시다.
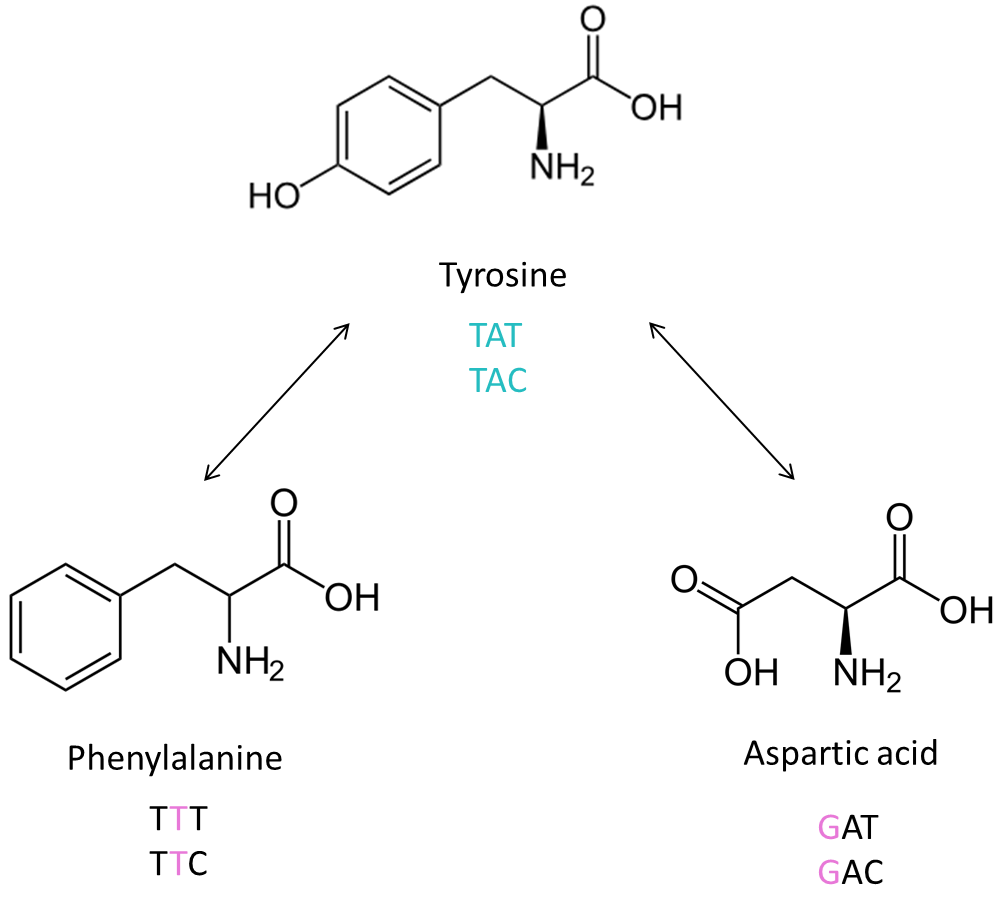
Phenylalanine, Aspartic acid는 Tyrosine과 오직 하나의 염기 차이를 보이지만, 구조적으로 유사한 Tyrosine과 Phenylalanine과 달리 Aspartic acid와는 큰 차이가 존재합니다.
이러한 아미노산 사이의 관계를 점수로 나타낸 행렬이 바로 치환 행렬(Substitution matrix 혹은 scoring matrix)입니다.
즉, 치환행렬은 아미노산의 진화적 관계(보존과 비보존)를 반영한 점수 표입니다. 단순한 문자 비교가 아닌, 단백질의 기능적 및 구조적 특성을 포함한 복잡한 정보를 바탕으로 점수를 매깁니다.
2) PAM matrices

최초로 substitution matrix를 제안한 사람은 Margaret Dayhoff입니다. Dayhoff와 동료들은 1978년에 scoring matrix인 Accepted point mutation(PAM) matrices을 개발했습니다. Dayhoff는 단백질 서열 간 치환 패턴을 연구하여 진화적 변화를 정량화한 인물로 "생물정보학의 어머니"로 불립니다.
Dayhoff는 단백질에 따라 진화속도가 다르다는 결과를 발표했습니다. 예를 들어, 히스톤은 1억 년 동안 아주 조금만 변했지만, 카파 카세인의 경우 히스톤보다 약 230배 빠르게 변했습니다.

Dayhoff 는 이 데이터를 기반 삼아 만들어진 아미노산 사이의 치환 및 보존 값을 PAM matrices로 나타내었습니다.
정확히 말하자면 PAM 행렬은 유사도(identity)가 85% 이상인 단백질 서열 그룹(70개 그룹)에서 아미노산 치환 패턴을 분석한 결과를 말합니다.
- PAM1: 100개의 아미노산에서 한 개의 아미노산이 치환될 확률을 나타냅니다 (1% divergence).
- PAM100: 100개의 아미노산 당 100번 치환이 일어날 확률을 나타냅니다. 그러나 모든 아미노산이 치환될 확률이 아니며, 이때 여러 번 치환으로 원래 아미노산으로 되돌아갈 수도 있음을 고려해야 합니다.
- PAM250: 100개의 아미노산에서 250번의 치환이 일어날 확률을 나타냅니다.
PAM250
가장 대표적인 matrix는 PAM250를 살펴봅시다.
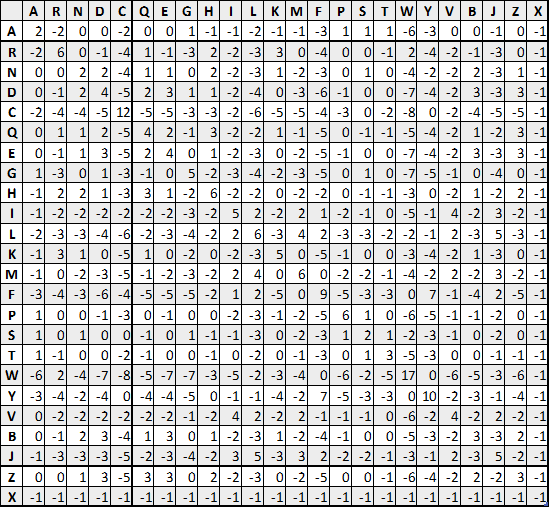
PAM matrix의 기본 가정은 "한 단백질 내에서의 특정 위치의 아미노산의 변화는 다른 위치의 아미노산들에 의해 영향받지 않으며, 또한 특정 위치에서 아미노산이 교체될 확률은 그의 위치와는 무관하다"는 것입니다. 그러나 이러한 가정은 생물학적으로 위배되었으며, 이에 대한 한계를 가졌다고 볼 수 있습니다.
또한 PAM은 글로벌정렬(Global aligmnent)을 기반으로 하며, 서열 간 진화적 거리가 클수록 신뢰도가 낮아지는 한계도 존재합니다.
3) BLOSUM matrices
BLOSUM(BLOcks SUbstitution Matrix)은 1991년에 Steven Henikoff Altschul과 Jorja Henikoff에 의해 제안된 치환행렬입니다. 이는 진화 모델에 기반하지 않고 실제 데이터 (BLOCKs database)의 관찰을 통해 만들어졌습니다. BLOCKs database는 아미노산 서열 중 conserved 한 지역의 데이터를 담고 있는 데이터베이스입니다.
또한 BLAST도 초기에는 PAM 행렬을 사용하였지만, 이제는 BLOSUM 행렬을 사용하고 있습니다.
BLOSUM도 PAM처럼 여러 버전에 존재하며, 적힌 숫자는 계산에 사용된 서열들의 보존(유사성)을 나타 냅니다
- BLOSUM62: 서열이 적어도 62% 동일할 때 아미노산이 쌍을 이룰 확률 계산하여 구한 행렬값
- BLOSUM45: 서열이 적어도 45% 동일할 때 아미노산이 쌍을 이룰 확률 계산하여 구한 행렬값
- BLOSUM90: 서열이 적어도 90% 동일할 때 아미노산이 쌍을 이룰 확률 계산하여 구한 행렬값

PAM과 BLOSUM의 비교
| PAM | BLOSUM |
| global alignments 를 기반으로 한다 | local alignments 를 기반으로 한다 |
| PAM1은 차이가 1% 이하이지만 identity 99%에 해당하는 서열의 비교로부터 계산된 행렬값이다. | BLOSUM 62는 62% 이하의 pairwise identity 을 갖는 서열의 비교로부터 계산된 행렬값이다. |
| score가 높을 수록 진화적 거리가 더 크다는 뜻 | score가 클 수록 유사성이 높고 진화적 거리가 짧다는 뜻 |
(https://en.wikipedia.org/wiki/BLOSUM#The_relationship_between_PAM_and_BLOSUM)
4. BLAST
BLAST(Basic Local Alignment Search Tool)는 1990년 미국 NCBI에서 개발된 유사성 검색 프로그램으로, 대규모 데이터베이스에서 서열의 유사성을 빠르게 검색할 수 있는 도구입니다. 이는 heuristic search algorithm와 Karlin-Altschuls의 통계방법을 채택하고 있습니다.
BLAST의 특징은 1) neighborhood words(확장 단어)을 도입하며 검색 효율을 높이고 2) Heuristic Search Algorithm을 사용해 속도를 높였으며, 3) BLOSUM 행렬을 기반으로 유사성을 점수화합니다.
이는 비슷한 방식의 FASTA에 비해 통계적으로 엄격하고, 더 sensitive 하다고 알려져 있습니다.
이제 BLAST의 작동 원리를 알아봅시다.
1) Word search
Query 서열에서 길이 w에 해당하는 작은 조각(Word)을 생성합니다. BLAST는 이를 데이터베이스 서열과 비교하여 빠르게 매칭을 수행합니다.
왜 word를 생성할까요? 이는 서열을 하나씩 비교하는 것보다 훨씬 빠르기 때문입니다. 이때 아미노산 서열 정렬에서 기본 word의 길이는 3, 염기에서는 11의 길이를 가집니다.
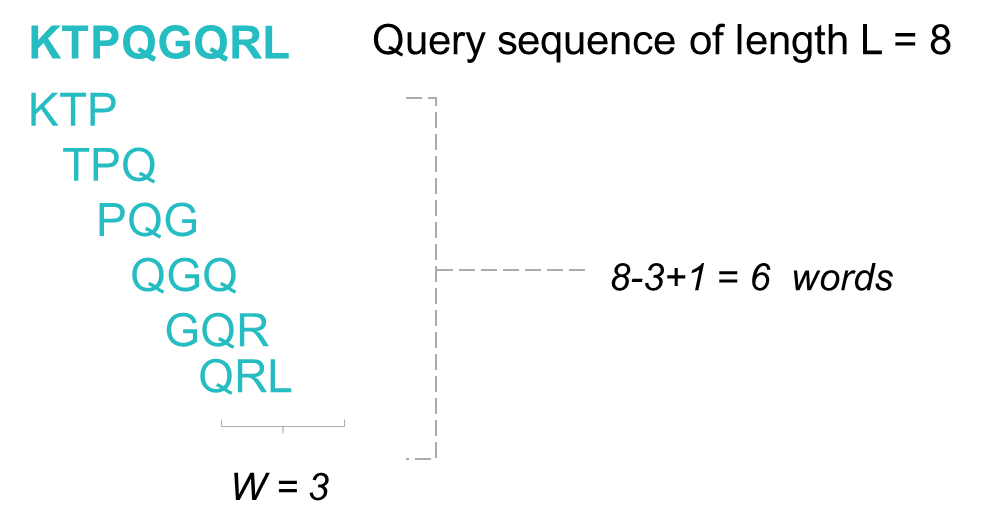
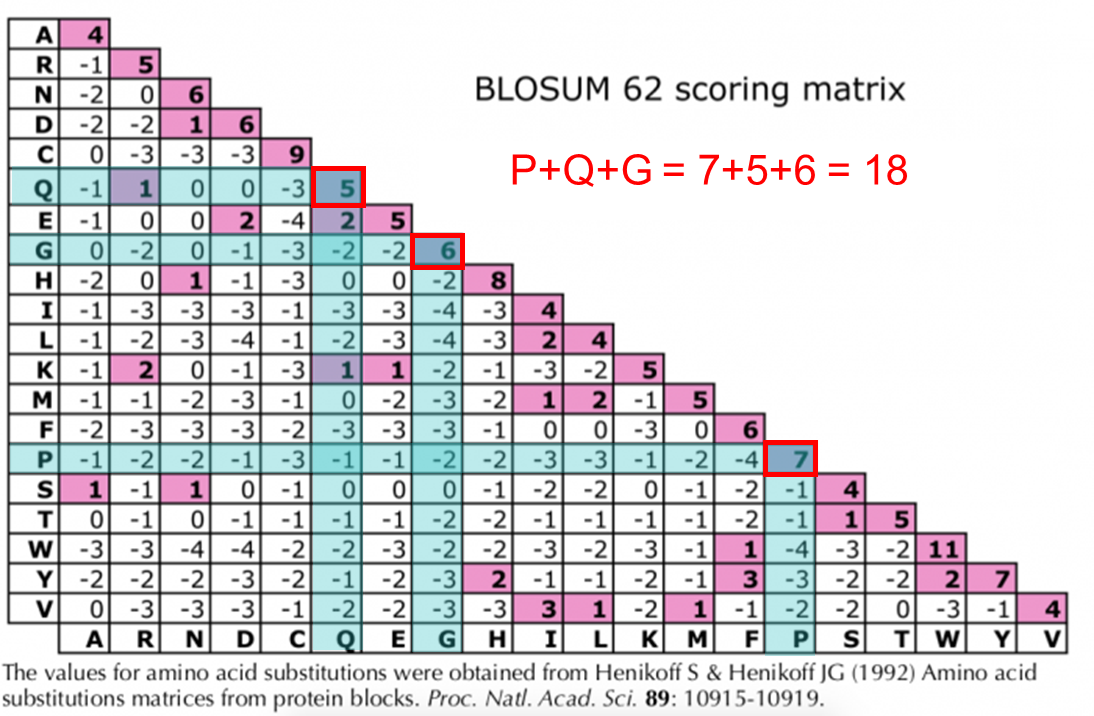
여러 word 중 PQG에 집중해 봅시다. PQG 단백질 서열이 얼마나 잘 보존되는지 알아보기 위해 BLOSUM 62 치환행렬을 기반으로 확인해야 합니다. BLOSUM 행렬에서 PQG의 보존 값은 18입니다.
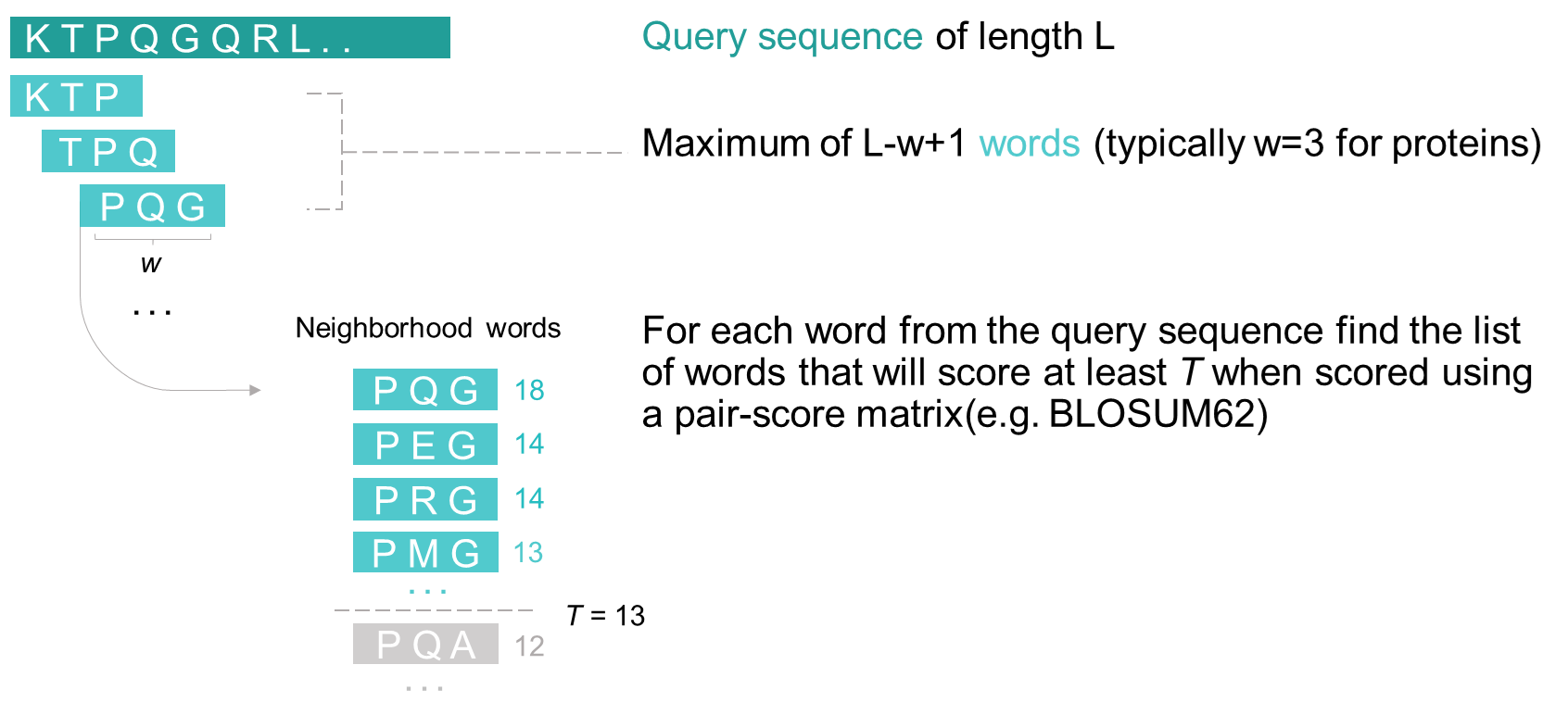
이때 우리는 BLOSUM 행렬 값을 기반으로 비슷한 단백질 서열들을 나열할 수 있습니다.
BLOSUM의 총 합(T) 값이 13 이상인 단백질 서열을 PQG와 유사한 word라고 정의해 봅시다.
이때 유사한 word 들을 Neighborhood words라고 합니다.
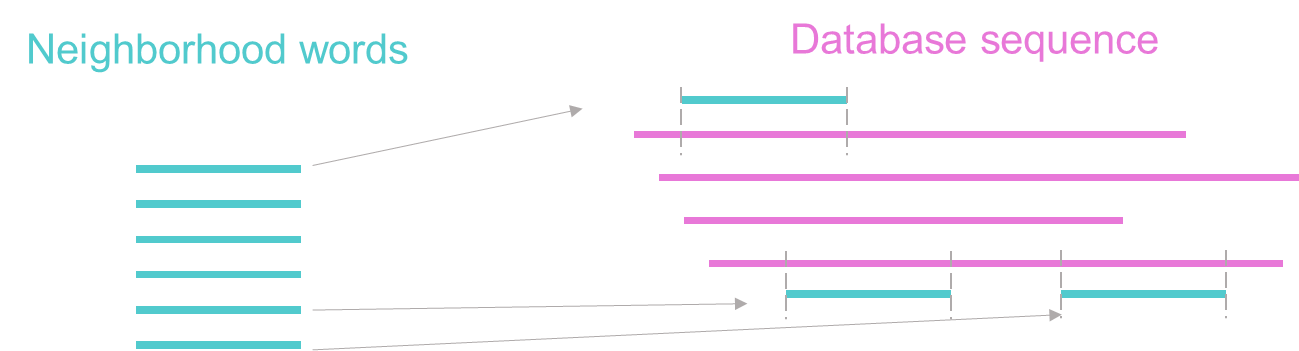
2) Identification of exact word match method
이제 Neighborhood words를 모두 알아보았다면, 이 단어 목록을 기준으로 database 서열과 일치하는지 알아봅니다. 이때, word와 database가 일치되는 부분을 seed라고 합니다
3) High segment pair (HSPs) alignment method
일치하는 Word(=seed)를 기준으로 양쪽으로 확장하여 유사성( Similarity )을 최대로 유지하는 지점까지 정렬합니다. Gap Penalty를 적용해 불필요한 간격을 최소화합니다.
- 유사성은 Similarity score (raw score)로 측정됩니다.
- Similarity score는 BLOSUM을 기반으로 각 염기들의 관계에 따라 match 혹은 mismatch 점수가 부과된 결과입니다.
- Seed를 기준으로 전체적인 Similarity score가 최대가 되는 지점(high scoeing pairs; HSP)까지 확장해 나갑니다.
seed를 방향으로 양쪽으로 확장해 나가면서 최대 유사도를 확인하는 것이 기본 알고리즘입니다. 그러나 이해를 돕기 위해 한쪽 방향으로 확장하면서 최대 유사도를 구하는 방법을 알아봅시다.
PQG-PMG 쌍에서 한쪽 방향으로만 유사한 서열을 확인한다고 할 때, 어디까지 확장해야 할까요?
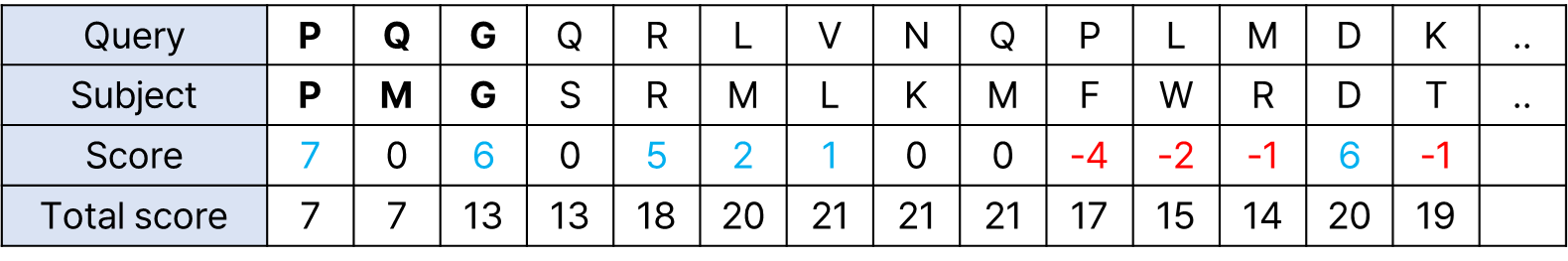
BLOSUM62를 기반으로 한 Similarity score는 아래와 같은 추세를 보입니다.
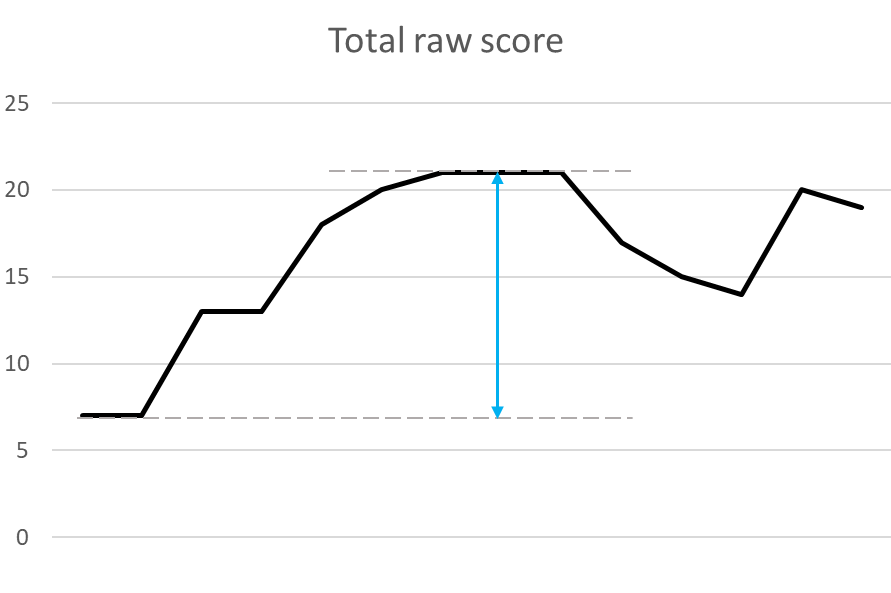
여기서 시작점부터, 상승 추세가 꺾이는 부분까지를 High score라고 판단하게 됩니다.
Gap penalty
정렬하려는 서열 사이에 Gap 은 어떻게 계산해야 할까요? 정렬하려는 두 서열사이에 Gap 이 있다면 이는 유사성이 떨어지는 것으로 판단해야 합니다. 즉, 페널티를 부과해야 합니다.
그렇다면 페널티를 모든 gap 한 칸 마다를 부과해야 할까요? 아니면 연속된 Gap 이 많을수록 더 많은 페널티를 부과해야 할까요?
아래 예시를 살펴보겠습니다. 아래 그림에는 동일하게 3개의 Gap이 존재합니다.
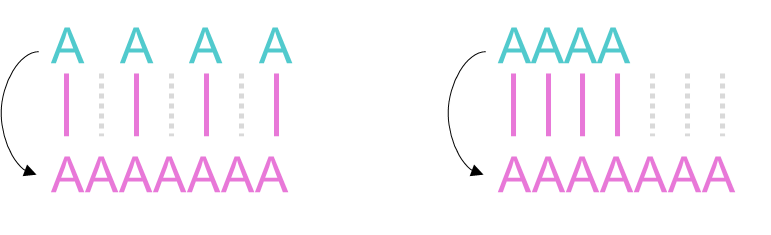
왼쪽은 각각 3개의 진화적 사건(event)으로 인해 Gap이 생성되었으며, 오른쪽은 하나의 사건로 인해 생성될 수 있는 Gap이라고 생각됩니다.
> 즉, 서열 정렬에서는 왼쪽에 더 많은 페널티를 부과해야 한다.
그렇다면 패털티는 어떻게 부과해야 이를 더 잘 반영할 수 있을까요?
> 이를 위해서는 Gap의 생성과 연장 모두에 페널티를 부과하는 것이 바람직합니다.
> BLAST에서는 Gap open에 -11점을, Gap extend에 -1을 부과하고 있습니다.
실제로 Gap penalty를 고려한 similarity score를 살펴봅시다.

High segment pair (HSP)에 대한 similarity score(S)는 match 시 점수(60) + mismatch 시 점수(-13) + gap시작에 대한 페널티(-13) + gap 연장에 대한 페널티(-13)를 모두 더한 값 (=35)입니다.
초기 BLAST 1.0에서는 GAP을 허용하지 않았습니다. 이는 대규모 데이터베이스 검색에 맞게 검색 속도를 최적화하기 위함입니다. 그러나 생물학적으로 Gap 은 유전체의 삽입 및 결실을 반영합니다. 또한 Gap 이 큰 서열은 데이터베이스 검색 후보군에서 제외할 수 있다는 장점도 있고, 컴퓨터의 계산성능이 늘어나면서 BLAST 2.0에서는 Gap을 고려한 결과를 나타내고 있습니다.
5. BLAST statistical significance
BLAST 결과를 판단할 때 고려하는 통계값이 어떻게 계산되는지 알아봅시다.
1) E-value
E-value는 기댓값(expected value)을 말하며, 두 서열의 유사성 점수(similarity score; S)가 특정 값 이상일 확률을 기반으로 우연히 나타날 수 있는 정렬의 수를 나타낸다.
즉, E-value 은 query마다 우연히 S값 이상의 정렬된 서열을 찾을 경우 hit 수에 대한 기댓값입니다.
E-value는 확률이 아니라, 기대되는 횟수라는 점을 기억해야 합니다.
길이 m과 길이 n을 가진 두 서열이 S이상의 score를 나타낼 기댓값(E value)sms Karlin-Altschul 방정식을 통해 계산됩니다.
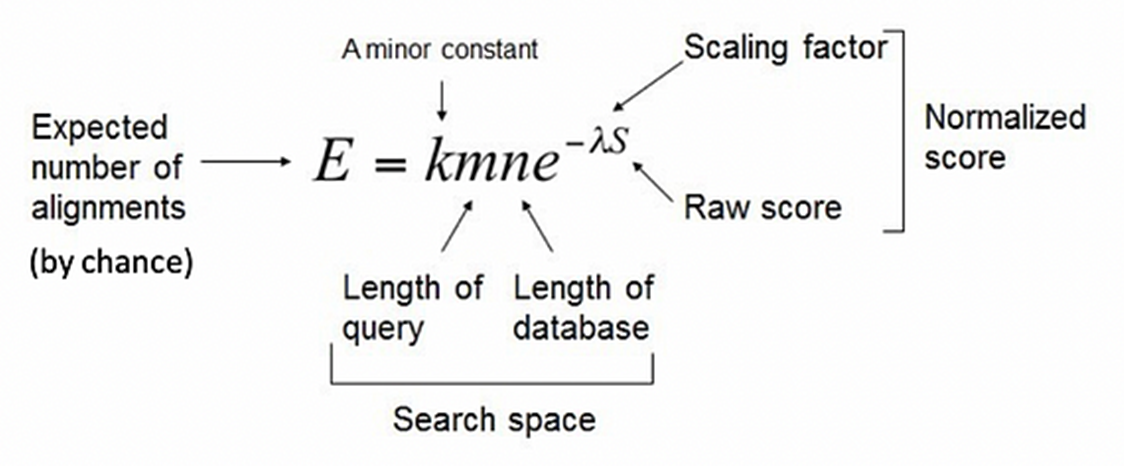
- m: Query 서열의 길이
- n: 데이터베이스 서열의 길이
- K: 검색 공간(Search Space)과 관련된 매개변수 (m과 n의 영향받음)
- λ: scoring system과 관련된 매개변수
- S: 정렬에 대한 유사도 점수(similarity score)
E-value의 특성
- m과 n의 값이 클수록: 데이터베이스와 Query가 길어지면 HSP(High Scoring Pair)가 우연히 나타날 확률이 증가하므로 E-value가 커집니다.
- S값이 클수록: 높은 점수를 가진 HSP는 더 유의미한 정렬을 나타내며, E-value는 지수적으로 감소합니다.
즉, E값은 0에 가까울수록 발견된 HSP보다 더 높은 값의 HSP 점수가 나타나지 않을 것을 뜻합니다.
E-value 해석하기
- E-value가 클수록: 우연히 정렬이 발생할 가능성이 높아져서 의미가 떨어집니다.
- E-value가 작을수록: 정렬이 우연이 아닐 가능성이 높아지고, 생물학적 의미가 있을 가능성이 큽니다.
| E-value | description |
| > 10^-1 | insignificant |
| 10^-5~10^-1 | distantly homologous |
| 10^-50 10^-5 | homologous proteins |
| 10^-100~10^-50 | nearly identical |
| <10^-100 | identical proteins |
(https://www.youtube.com/watch?v=S3gr8gjKHhc&t=2s)
예를 들어, E-value가 0.01이라면 100번의 검색 중 1번은 우연히 발생할 수 있다는 의미입니다. 따라서 데이터베이스가 커질수록 낮은 E-value가 중요합니다. (https://www.researchgate.net/post/What_is_E-value_exactly_means_and_what_is_it_1e-63_representing)
2) Bit score
Bit Score는 BLAST의 Raw Score(S)를 정규화한 값입니다. Raw Score(S)는 비교하는 서열의 길이와 치환행렬(BLOSUM)에 따라 달라지는데, 이를 보정하기 위해 Bit Score (S')가 사용됩니다.
즉, K, λ 값은 HSP 마다 다르기 때문에 다른 서열과의 비교를 위해 S값을 K, λ로 정규화 해줍니다.
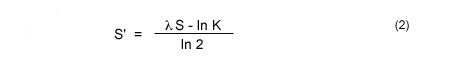
Bit Score는 서로 다른 BLAST 결과를 비교할 때 중요한 기준이 됩니다. 높은 Bit Score는 더 신뢰할 수 있는 정렬을 나타냅니다.
E-value와 Bit Score 관계
Bit Score를 사용해 E-value를 다시 계산할 수 있습니다. 이를 보면 E-value값에서 중요한 변수는 m과 n에 따른 검색공간(search space)이 임을 다시 한번 확인할 수 있습니다.
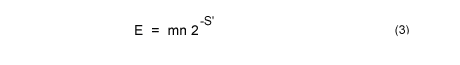
만약
λ = 1.37,
S = 23,
k = 0.771일 때,
Bitscore의 값은 약 46입니다.
또한 m이 34,
n이 5,854,611,841이라고 가정할 때,
E-value는 34*5854611841*2^(-46) = 0.003입니다.
3) P-value
P-value는 통계적으로 관찰된 점수 HSP가 우연히 발생할 확률을 나타냅니다. P-value는 Gumbel Extreme Value Distribution(EVD)을 기반으로 계산되며, E-value와 밀접한 관련이 있습니다.
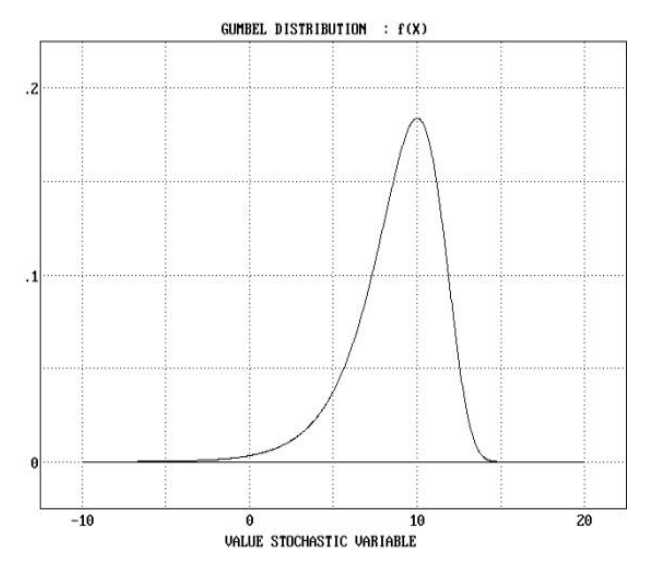
x보다 크거나 같은 점수 S를 관찰할 확률 p는 다음 방정식으로 제공된다.
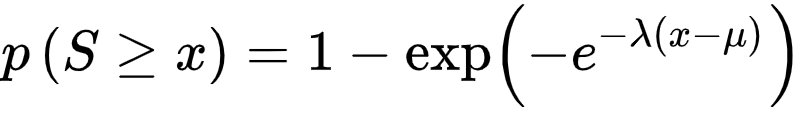
이때 μ값은 아래와 같다.
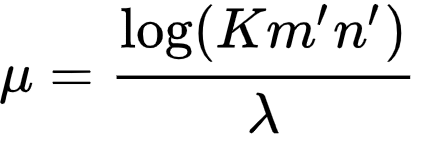
혹은 아래와 같이 표현 가능하다.
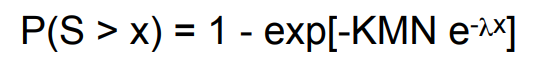
E-value가 매우 작다면 P-value 또한 작아지며, 두 값은 유사한 의미를 가집니다.
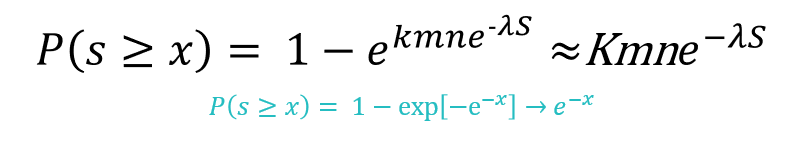
P-value와 E-value의 차이점
- E-value는 특정 점수 이상의 정렬 수를 기대하는 횟수.
- P-value는 특정 점수 이상이 우연히 관찰될 확률.
실제 BLAST 결과에서 E-value, P-valeu, Bit score를 계산해 보자 (Ref: ➕)
아래 결과는 Human p53 단백질을 Swiss-Prot database(C.elegans) 데이터베이스에서 에서 정렬한 결과이다.
- Query 서열의 길이 = 305
- 데이터베이스 서열의 길이 = 1,414,501
- K = 0.0410
- λ = 0.267
- Similarity score(S): 77
Q. 이때의 Bit score와 E-value는?
Bit score 계산:
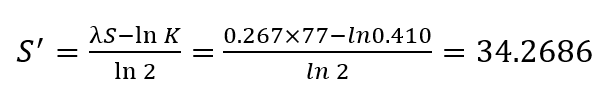
E-value 계산:
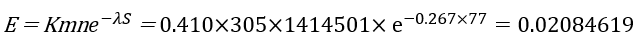
Q. 이때 p-value 값은?
P-value 계산:
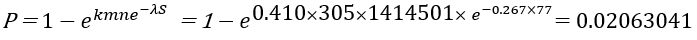
결과적으로 E value가 작으면, P-value값과 유사하게 나타납니다.
참고
BLAST Statistics
- https://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html 👍 👍 👍
- 한국어 강의 (https://www.youtube.com/watch?v=c7xqId2Sto0)
BLAST algorithm
- blast용어: https://www.ncbi.nlm.nih.gov/books/NBK62051/
- https://www.youtube.com/watch?v=wfi_KimrNQM
- MIT open course, Foundations Of Computational And Systems Biology, 2014 (https://ocw.mit.edu/courses/7-91j-foundations-of-computational-and-systems-biology-spring-2014/resources/lecture-2-local-alignment-blast-and-statistics/)
- RobEdwards. BLAST 2 seed and extend (https://www.youtube.com/watch?v=kAAme1fBanc)
+) 요약 강의: https://www.youtube.com/watch?v=g0nSH17psDc 👍 👍
- http://ibi.zju.edu.cn/bioinplant/temp/BSA/BSA_Blast_BLAST.pdf
- 유재천 외. (2022). 유전자 발굴 알고리즘 개발, 한국과학기술정보연구원
- Mount DM. (2004). Bioinformatics: Sequence and Genome Analysis. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY. ISBN 978-0-87969-608-5.
Alignment
-Altschul SF. (1991). Amino acid substitution matrices from an information theoretic perspective. Journal of Molecular Biology. doi:10.1016/0022-2836(91)90193-A
- Weik TC, Bio Informatics (https://www.fh-muenster.de/eti/labore_forschung/db/bio-informatics.php) 👍
PAM
- Dayhoff MO, Schwartz RM, Orcutt BC. (1978). A model of Evolutionary Change in Proteins. In: Atlas of protein sequence and structure. National Biomedical Research Foundation. Washington, DC. ISBN 978-0-912466-07-1.