
작성 날짜: 2023.10.11. 수

| 글 작성에 앞서..
shotgun metagenome 분석은 확실히 amplicon 보다 어려운데 16S full-length 분석이 어려운가? 아니다 amplicon이랑 동일하다! 위 글도 amplicon 데이터 분석을 모두 파악하고 있다는 가정 하에 작성하였다.
| 개요
▶ Dada2란? R을 기반으로 qiime2(리눅스 기반)와 같이 미생물 분석에 사용되는 R 패키지이다.
▶ 튜토리얼 목표 : Dada2로 박테리아의 16S full-length 데이터의 처리와 phyloseq을 통한 시각화
▶ 지난 글 참고
- Dada2 16S amplicon 분석: https://bio-kcs.tistory.com/entry/R-Dada2-%ED%8A%9C%ED%86%A0%EB%A6%AC%EC%96%BC-ver-11
- Dada2 ITS 분석: https://bio-kcs.tistory.com/entry/DADA2-Dada2-ITS-Tutorial
| long read data의 생산 방법
현재 가장 많이 사용되는 롱리드 시퀀싱 방법으로는 pacbio사의 SMRT와 oxford사의 nanopore가 존재한다.
Pacbio사의 SMRT(Single Molecule, Real-Time) 시퀀싱
- single DNA polymerase 가 각 칸의 바닥에 붙어있는 Zero-mode waveguide (ZMW)를 이용한 시퀀싱 빵법이다.
- Zero-mode waveguide 한 Michael J. Levene 외 연구팀이 개발한 단일 분자 분석법으로, 작은 칸 안에 하나의 구성물질만 들어가게 하고, 그 칸 안에서만 형광분자를 관찰할 수 있게 만든 구조로, 단일 분자 연구를 가능하게 한다.
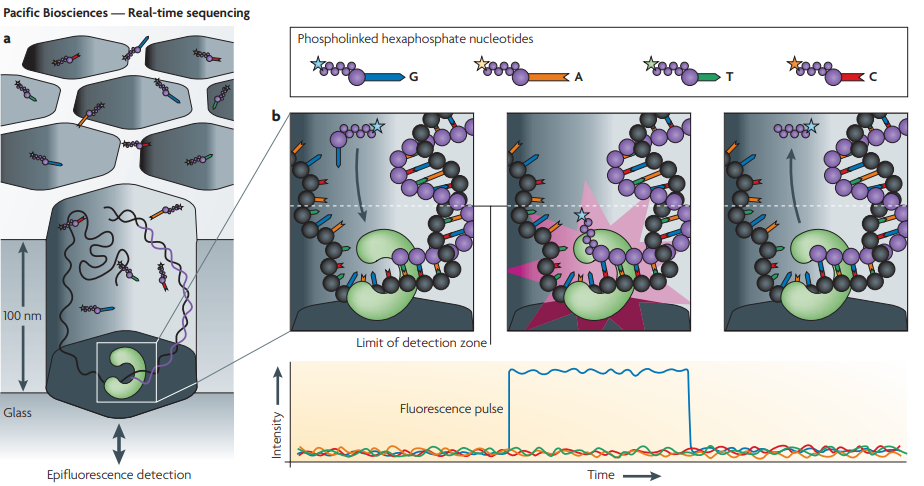
- 일루미나 시퀀서와 마찬가지로 형광분자를 타깃으로 탐지한다.
- 단점은 single strand만 읽어내며, 시그널이 높지 않으면 판별하기 어렵다.
Oxford 사의 nanopore 시퀀싱
oxford사에서도 롱리드 시퀀서를 개발하고 있던 와중에, 형광분자를 사용한 방법이 개발되었다. 아마 이에 대한 특허 분쟁을 피하기 위해 Oxfold사는 다른 방법으로 염기를 판별하는 기술을 개발했을 것이다.

- 뉴클레오 타이드의 크기에 따른 port 크기의 차이 때문에 이온의 흐름을 방해하는 정도가 다르다. 이에 따른 이온이 이동 통과 시간, 전류 강하, 진폭으로 염기를 구분한다. SMRT보다 더 다양한 염기를 구분 가능하다.
- nanopore의 가장 큰 장점은 시퀀싱 기기가 이동이 가능하다는 점이다. 그러나 이 기기에 있는 데이터를 해석하는 컴퓨터가 상당히 고가라고 알려져 있으며, 생물학적 분자를 사용하기 때문에 사용기한이 정해져 있다.
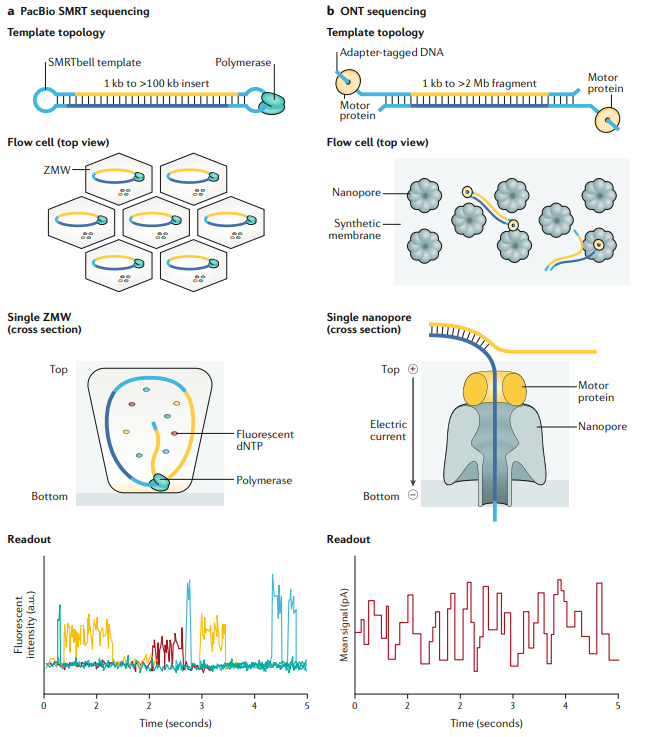
두 방법을 위 그림으로 간단하게 비교할 수 있다. a. 는 각 칸에 single DNA 분자를 넣어서, DNA polymerase가 합성을 해가면서 형광분자를 탐지한다. b nanoport는 pore에서 각 분자가 합성될 째 전류를 탐지해서 염기를 읽어드린다.
| Amplicon data와 16S full-length의 차이

박테리아의 16S rRNA는 위 그림과 같이 약 9개의 변이가 다양한 부분이 존재합니다. 이러한 다양성 때문에 다양한 박테이라의 계통을 구분하는 데에 활용할 수 있습니다. 장마이크로바이옴은 대부분 V4, V3-V4, V4-V5등을 사용하고, 피부마이크로바이옴은 V1-V2, V1-V3를 연구에 활용합니다.

위 그림은 여러 amplicon 영역을 사용하여 계통수를 확인했을 때 unclassified로 나온 계통을 빨간색으로 나타낸 결과입니다. 이를 보면 16S를 사용할 때, V1-V9영역을 모두 사용한 것이 unclassified의 비율을 줄일 수 있는 확실한 방법입니다.
| DADA2를 제외한 분석법이 있는가?
- 16S rRNA full-length를 분석하는 대에 사용하는 도구는 QIIME, DADA2, Mothur, MEGAN가 있다고 한다.
- DADA2를 이용한 16S full-length 분석법에 대한 논문의 인용수는 338회(2023.10.11 기준)로 다른 방법에 비해 인용수가 많다. QIIME2를 사용하는 사람들도 결국 DADA2의 denoise방법을 사용하기 때문에, 분석 알고리즘은 dada2를 가장 많이 사용할 것으로 보인다.
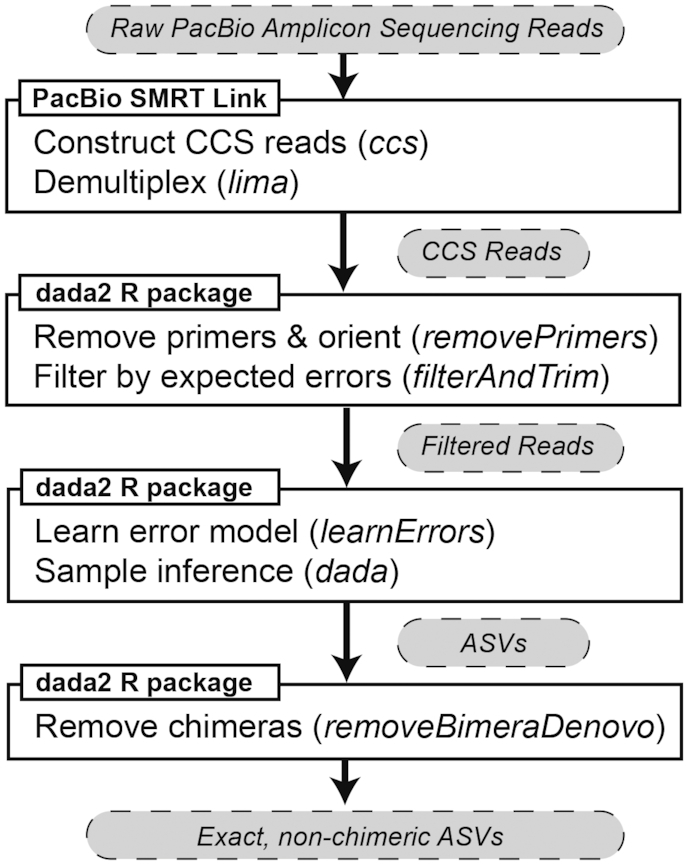
- 생물정보학 데이터를 대량으로 다루기 위해 개발된 Next flow를 사용한 분석 파이프라인도 존재한다. 개발된 지 오래되지 않아서 인용수는 매우 적은 편. 우리 연구실에서도 아직 16S full length에 대한 데이터가 크지 않아서 사용해보지는 않았다. 그러나 사용해 보면 확실히 편리할 것으로 예상된다.

만약 당신이 시퀀싱 회사에 샘플을 맞긴 후 raw data을 받게 되었다면, 파일의 형식은 HiFi(SMRT 시퀀싱일 때) 일 것이다.
일반적인 long read와 다르게 HiFi read는 원형의 DNA를 반복적으로 읽어드린 후, 이를 잘라낸 서열들을 말한다.


HiFi 데이터는 fastq.gz파일과 bam, bam.pdi, xml파일을 포함하고 있다. 보통 long read 시퀀싱이 single cell에 많이 사용되는데, 이때 유전체의 자리를 reference와 mapping 한 결과를 나타낸 BAM파일과 PacBio BAM index file인 bam.pbi가 같이 첨부 외어있으며, XML는 각 데이터에 대한 메타데이터가 포함되어 있다.
xml데이터를 txt로 열었더니 아래와 같은 정보를 확인할 수 있었다.

| Tutorial
1. 데이터 준비
- HiFi파일의 압축을 푼 후, fastq.gz파일을 준비한다.
- Cutadapt단계를 생략하고 dada2를 사용하여 primer만 잘라낼 수 있다. 물론 cutadapt를 사용해도 문제는 없다!
2. 준비된 파일을 R에서 불러오자
library(dada2);packageVersion("dada2")
## [1] '1.12.1'
library(Biostrings); packageVersion("Biostrings")
## [1] '2.46.0'
library(ShortRead); packageVersion("ShortRead")
## [1] '1.36.1'
library(ggplot2); packageVersion("ggplot2")
## [1] '3.1.0'
library(reshape2); packageVersion("reshape2")
## [1] '1.4.3'
library(gridExtra); packageVersion("gridExtra")
## [1] '2.3'
library(phyloseq); packageVersion("phyloseq")
## [1] ‘1.42.0’
library(dplyr); packageVersion("dplyr")
## [1] ‘1.0.10’
# 미리 경로 지정하기
path.out <- "~/02.dada2/Figures/"
path.rds <- "~/02.dada2/RDS/"
rc <- dada2:::rc
path <- "~/01.data" # 본인의 분석 경로에 따라 변경
path.remove_primer <- "~/02.dada2/Removeprimer"
list.files(path)
3. 퀄리티를 확인해 보자
fn <- sort(list.files(path, pattern = "_HiFi.fastq.gz", full.names = TRUE))
sample.names <- sapply(strsplit(basename(fn), "_"), `[`, 1)
plotQualityProfile(fn[1:2])
4. primer를 제거해 보자
- 16S full-length는 보통 V1-V9을 읽어냄으로 대부분 같은 primer를 사용한다.
- 아래 코드는 DADA2 ITS tutorial 에 포함되어있으며, 각 read에 붙어있는 primer의 개수를 찾는다.
- 너무 적다면 primer가 올바른지 확인해 봐야 하며, 붙은 위치가 이상하다면, primer를 reverse로 바꾸어서 사용해봐야 한다.
# dada2 totorial primer
F27 <- "AGRGTTYGATYMTGGCTCAG"
R1492 <- "RGYTACCTTGTTACGACTT"
allOrients <- function(primer) {
# Create all orientations of the input sequence
require(Biostrings)
dna <- DNAString(primer) # The Biostrings works w/ DNAString objects rather than character vectors
orients <- c(Forward = dna, Complement = Biostrings::complement(dna), Reverse = reverse(dna),
RevComp = Biostrings::reverseComplement(dna))
return(sapply(orients, toString)) # Convert back to character vector
}
primerHits <- function(primer, fn) {
# Counts number of reads in which the primer is found
nhits <- vcountPattern(primer, sread(readFastq(fn)), fixed = FALSE)
return(sum(nhits > 0))
}
FWD.orients <- allOrients(F27)
REV.orients <- allOrients(R1492)
rbind(FWD.ForwardReads = sapply(FWD.orients, primerHits, fn = fn[[1]]),
FWD.ReverseReads = sapply(FWD.orients, primerHits, fn = fn[[1]]),
REV.ForwardReads = sapply(REV.orients, primerHits, fn = fn[[1]]),
REV.ReverseReads = sapply(REV.orients, primerHits, fn = fn[[1]]))샘플에 프라이머를 매칭해 보니, 잘 매칭된 것을 할 수 있다.
DADA2 자체에서 프라이머를 제거해주자
noprimer <- file.path(path.remove_primer, basename(fn))
prim2 <- removePrimers(fn, noprimer, primer.fwd=F27, primer.rev=dada2:::rc(R1492), orient=TRUE)
5. 각 read의 길이를 확인해 보자
lens.fn <- lapply(noprimer, function(fn) nchar(getSequences(fn)))
lens <- do.call(c, lens.fn)
hist(lens,breaks = 100)대부분 1400~1500대일 것이다.
6. 전처리
# 퀄리티 필터링
filts2 <- file.path(path.remove_primer, "filtered", basename(fn))
track2 <- filterAndTrim(noprimer, filts2, minQ=3, minLen=1000, maxLen=1600, maxN=0, rm.phix=FALSE, maxEE=2) # default setting
track2
# 중복된 read 합치기
drp2 <- derepFastq(filts2, verbose=TRUE)
# 에러율 보기
err2 <- learnErrors(drp2, errorEstimationFunction=PacBioErrfun, BAND_SIZE=32, multithread=TRUE)
saveRDS(err2, file.path(path.rds, "err2.rds"))
# denoising = ASV 구하기
dd2 <- dada(drp2, err=err2, BAND_SIZE=32, multithread=TRUE)
saveRDS(dd2, file.path(path.rds, "dd2.rds"))위 옵션은 DADA2 tutorial에 기반하여 지정되었다.
7. 구해진 ASV의 taxonomy 정보 알아보기
# 전처리 과정에 따른 read수 확인
cbind(ccs=prim2[,1], primers=prim2[,2], filtered=track2[,2], denoised=sapply(dd2, function(x) sum(x$denoised)))
st2 <- makeSequenceTable(dd2); dim(st2)
# SILVA
SILVA_tax2 <- assignTaxonomy(st2, "~/silva_nr99_v138.1_train_set.fa.gz", multithread=TRUE) # Slowest part
SILVA_tax2[,"Genus"] <- gsub("Escherichia/Shigella", "Escherichia", SILVA_tax2[,"Genus"]) # Reformat to be compatible with other data sources
SILVA_tax2.sp <- addSpecies(SILVA_tax2, "~/silva_species_assignment_v138.1.fa.gz")
head(unname(SILVA_tax2.sp))
SILVA_tax2.sp %>% head
SILVA_tax2.sp %>% View
# 키메라 서열 제거
bim2 <- isBimeraDenovo(st2, minFoldParentOverAbundance=3.5, multithread=TRUE)
table(bim2)
# FALSE TRUE
# 455 1데이터베이스의 위치는 본인의 컴퓨터 경로에 따라 수정 바람
8. 키메라 서열 제거
seqtab.nochim <- removeBimeraDenovo(st2, method="consensus", multithread=TRUE, verbose=TRUE)
View(bim2)
sum(st2[,bim2])/sum(st2)
# [1] 0.0002543605키메라 서열이 없다면 에러가 뜰 것이다.
9. 계통수 만들기
library(DECIPHER); packageVersion("DECIPHER")
library(phangorn)
sequences <- getSequences(st2)
names(sequences) <- sequences
alignment <- AlignSeqs(DNAStringSet(sequences), anchor = NA)
phang.align <- phyDat(as(alignment, "matrix"), type="DNA")
dm <- dist.ml(phang.align)
treeNJ <- NJ (dm) # Note, tip order = sequence order
fit = pml(treeNJ, data=phang.align)
fitGTR <- update(fit, k=4, inv=0.2)
fitGTR <- optim.pml (fitGTR, model="GTR", optInv=TRUE, optGamma=TRUE,
rearrangement = "stochastic", control = pml.control(trace = 0)) # Time
saveRDS(fitGTR, file.path(path.rds, "tree.rds"))
fitGTR$tree
11. phyloseq 만들기
# phyloseq만들기
silva_ph <- phyloseq(otu_table(st2, taxa_are_rows=F),
sample_data(meta),
tax_table(SILVA_tax2.sp), phy_tree(fitGTR$tree))
# 계통수에 root설정하기
set.seed (42)
phy_tree(silva_ph) <- root(phy_tree (silva_ph), sample(taxa_names (silva_ph), 1), resolve.root = TRUE)
is.rooted(phy_tree(silva_ph)) # TRUE
# 저장
silva_ph
saveRDS(silva_ph, file.path(path.rds, "silva_phyloseq.rds"))여기서 meta데이터는 phyloseq의 sample data format에 맞추어 샘플의 데이터를 수정하면 된다. 대표적으로 meta data의 rownames은 각 샘플의 이름이 와야 한다. 또한 이 샘플 이름은 OTU table의 샘플 이름과 완전히 동일해야 한다.
완성된 phyloseq파일은. rds로 간편하게 저장하고, 다른 분석 환경에서도 readRDS()로 불러올 수 있다.
간단한 시각화
## alpha
alpha_value_table <- estimate_richness(silva_ph, measures=c("Observed", "Simpson", "Shannon", "Chao1"))
## Beta
silva_ph.ord <- ordinate(silva_ph, "PCoA", "wunifrac")
p1.SampleID = plot_ordination(silva_ph, silva_ph.ord, type="SampleID", color="SampleID", title="SampleID")
p1.SampleID
## taxonomy
silva_ph.rel <- transform_sample_counts(silva_ph, function(x) x / sum(x))
# 1) phyloseq 함수 사용
p <- plot_bar(silva_ph.rel, x="SampleID", fill="Phylum")
p
# 2) ggplot2 사용
melt <- psmelt(silva_ph.rel)
level = "Phylum"
x_ = "SampleID"
y_ = "Abundance"
tax_data = melt
plot <- ggplot(data=tax_data, aes_string(x=x_, y=y_, fill=level)) +
geom_bar(aes(), position="fill", stat="identity") +
labs(y = "Relative Abundance (%)", x = "") +
theme(axis.text.x.bottom = element_text(angle = 90, vjust = 0.4, hjust=1),
axis.text = element_text(size = 7),
legend.position = "right",
legend.background = element_rect(fill='transparent'),
legend.box.background = element_rect(fill='transparent', color=NA),
plot.background = element_rect(fill='transparent', color=NA),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_blank())
plot
12. taxonomy table 수정
- 참고: https://www.yanh.org/2021/01/01/microbiome-r/
| 참고
- Callahan BJ, Wong J, Heiner C, Oh S, Theriot CM, Gulati AS, McGill SK, Dougherty MK. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 2019 Oct 10;47(18):e103. doi: 10.1093/nar/gkz569. PMID: 31269198; PMCID: PMC6765137.
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010 Jan;11(1):31-46. doi: 10.1038/nrg2626. Epub 2009 Dec 8. PMID: 19997069.
- Logsdon GA, Vollger MR, Eichler EE. Long-read human genome sequencing and its applications. Nat Rev Genet. 2020 Oct;21(10):597-614. doi: 10.1038/s41576-020-0236-x. Epub 2020 Jun 5. PMID: 32504078; PMCID: PMC7877196.
- Johnson JS, Spakowicz DJ, Hong BY, Petersen LM, Demkowicz P, Chen L, Leopold SR, Hanson BM, Agresta HO, Gerstein M, Sodergren E, Weinstock GM. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019 Nov 6;10(1):5029. doi: 10.1038/s41467-019-13036-1. PMID: 31695033; PMCID: PMC6834636.
- Levene, M.J., et al., Zero-Mode Waveguides for Single-Molecule Analysis at High Concentrations. Science, 2003. 299(5607): p. 682-686.
- https://pacbiofileformats.readthedocs.io/en/11.0/DataSet.html
- https://en.wikipedia.org/wiki/Single-molecule_real-time_sequencing
- https://en.wikipedia.org/wiki/Zero-mode_waveguide
- https://github.com/PacificBiosciences/pb-16S-nf
- How nanopore sequencing works: https://www.youtube.com/watch?v=RcP85JHLmnI&t=7s
- PacBio Sequencing – How it Works: https://www.youtube.com/watch?v=_lD8JyAbwEo
- NGS데이터분석 제2강 파트 3 - Pacific Bioscience 시퀀싱 원리: https://www.youtube.com/watch?v=L98pLI5PMX0
- Pacbio data + DADA2: https://github.com/benjjneb/LRASManuscript
- https://benjjneb.github.io/LRASManuscript/LRASms_fecal.html
- Loopseq + DADA2: https://github.com/benjjneb/LoopManuscript