
글 작성: 24.01.03.
내용추가: 24.01.02.
1. Network analysis
| 마이크로바이옴 연구에서 네트워크 연구란?
- microbiome은 복잡한 미생물 군집을 말한다. 여러 생물들의 상호작용은 전체 미생물의 구조를 안정적이며 견고하게 만든다.
- 네트워크 plot은 미생물의 scale과 다양성을 모두 나타낼 수 있으며, 단편적인 관계보다 보편적인 시스템을 보여준다.
| Network의 구성성분
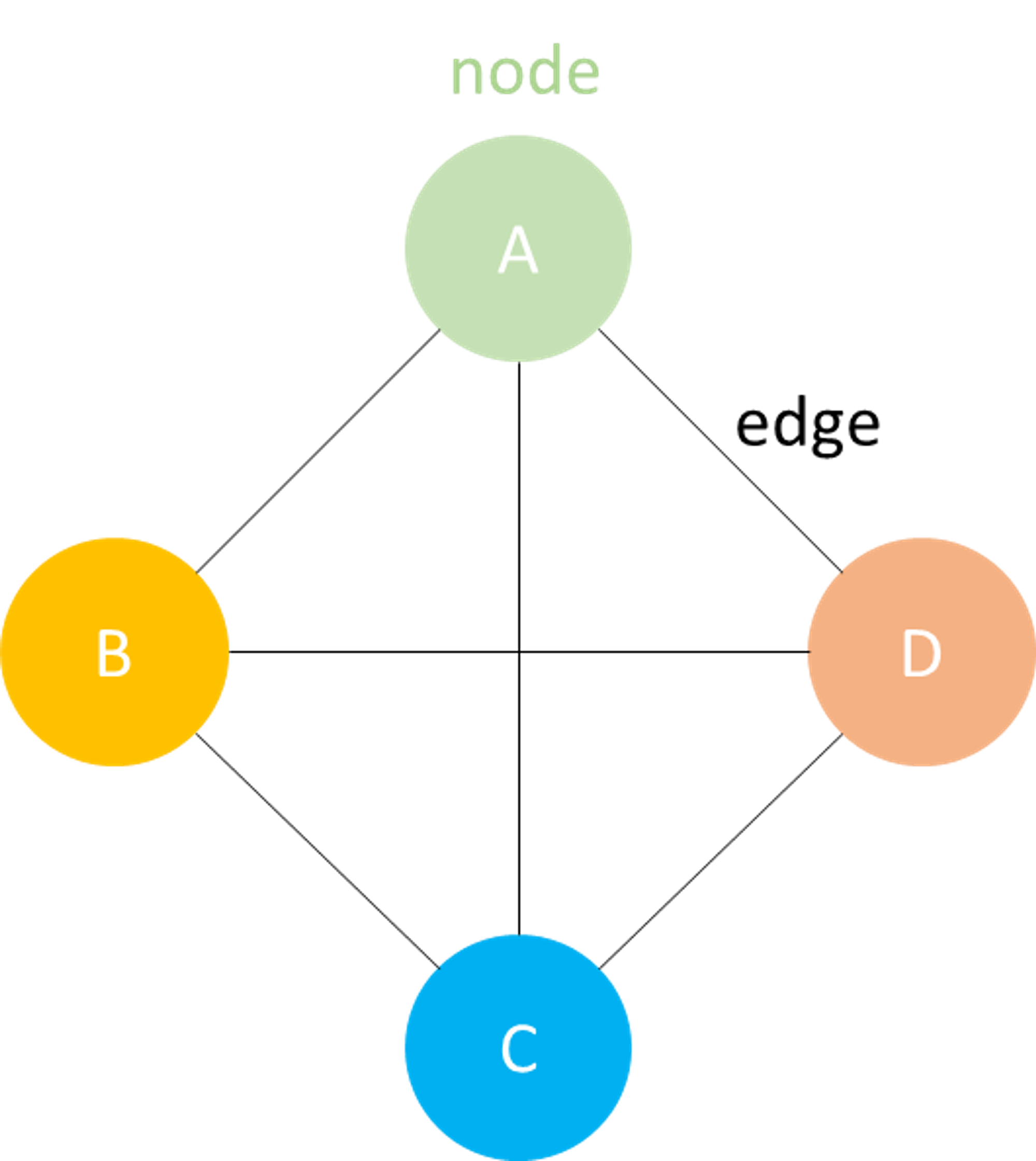
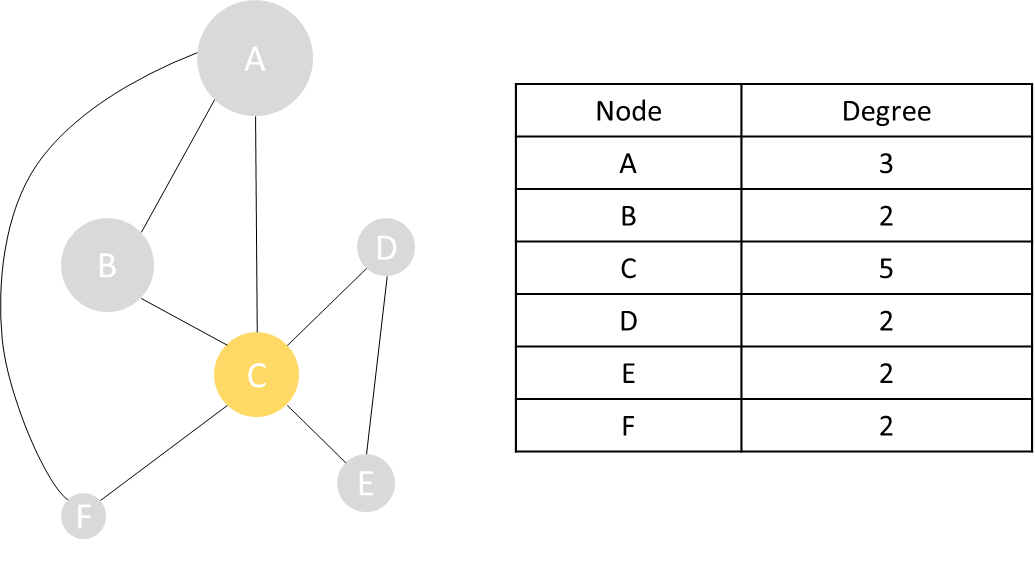
- node: 하나의 샘플이나 OTU/ASV를 뜻한다
- edge: 각 단위 간의 관계를 말한다
| network plot에서 알 수 있는 정보들
- node의 정보: 색, 모양, 크기, 라벨에 정보를 표시할 수 있다.
- e.g. 색에는 그룹 데이터를, 크기는 relative abundance를 나타낼 수 있다

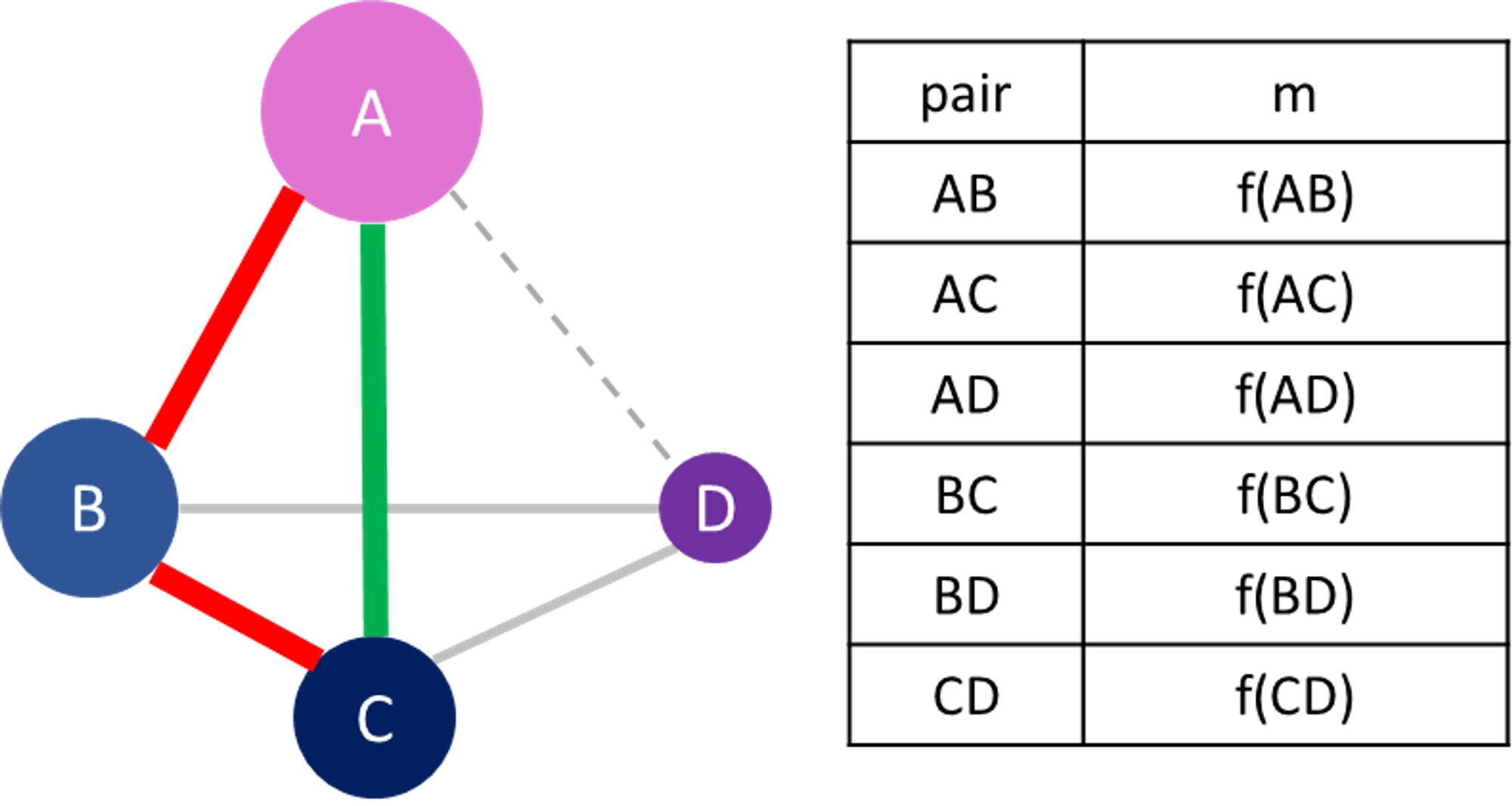
- Edge 정보 : dge의 두께, 색, 모양에 따라 정보를 표시할 수 있다.
- e.g. 빨간색은 음의 상관계수, 초록색은 양의 상관계수를 나타냄
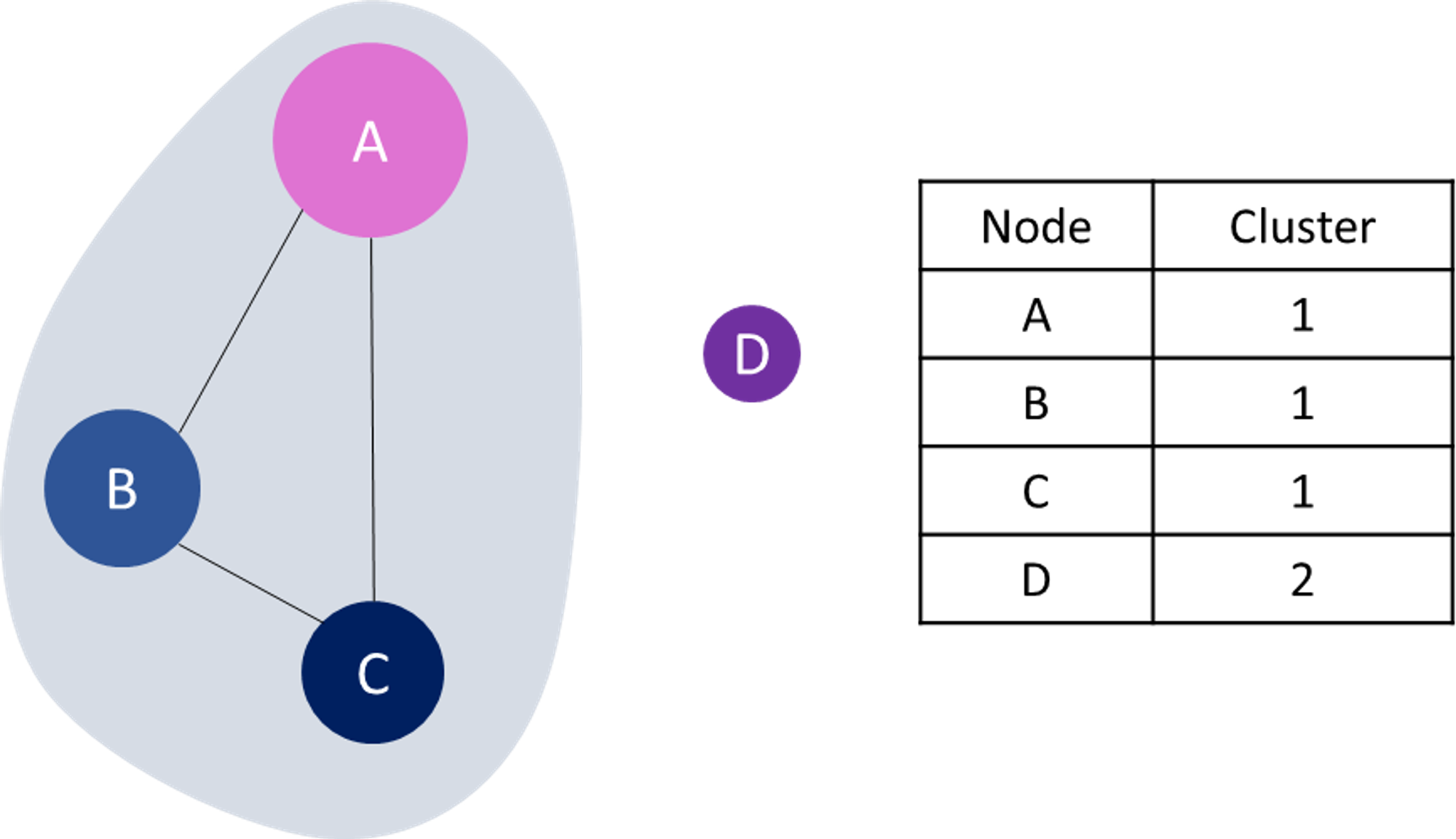
- 군집의 정보: 독립적인 생물군과 cluster를 구분 지을 수 있다
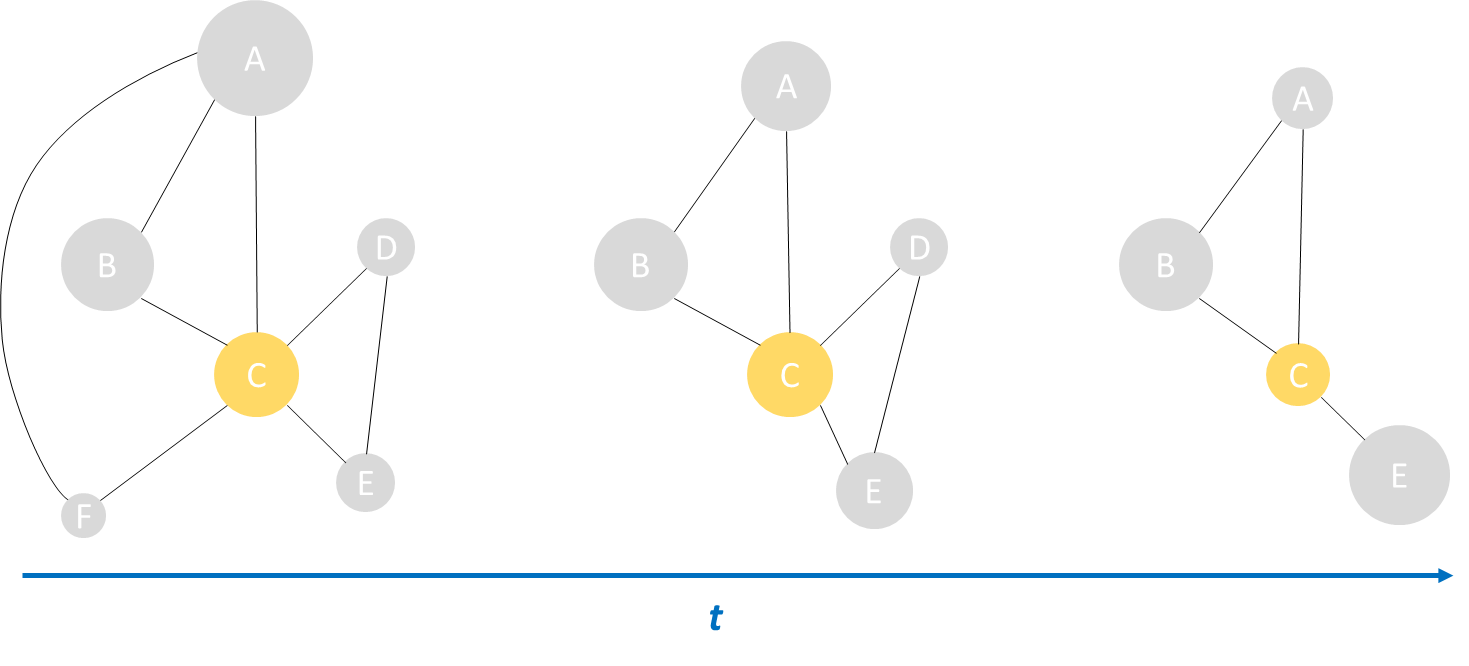
- Network Dynamic: 시간에 따라 네트워크 간의 전체 적인 연결 변화와 abundance변화를 관찰할 수 있다
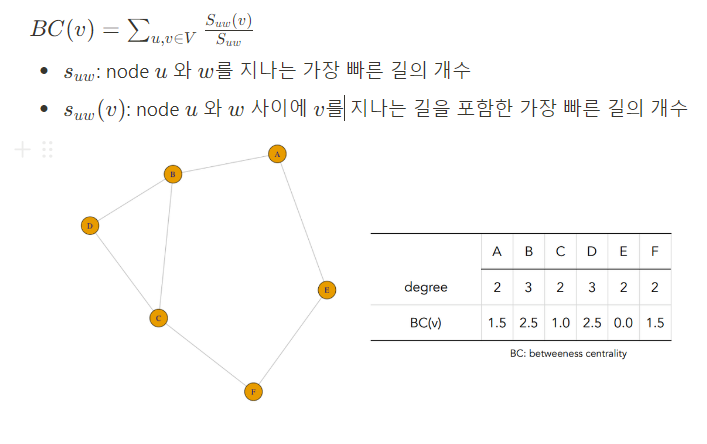
- Hub(Keystones) 정보: 전체 네트워크에서 상호작용이 가장 많은 key stone 생물을 찾을 수 있다. 이는 각 node의 Centrality를 측정하며, 측정 방법은 Degree, Closeness, Betweenness 등이 있다.
Betweenness centrality
| network 알고리즘
- Dissimilarity-Based Methods
- 가장 간단하고 빠른 방법으로, pairwise dissimilarity index(e.g. Bray-Cutris) 등을 이용하는 방법이다.
- phyloseq 패키지는 위 방법을 지원하고 있다.
- Correlation-Based Methods
- 상관관계 기반 방법은 Dissimilarity-Based의 대안으로 가장 인기 있는 방법이다. OTU 간의 pairwise 상호관계를 Pearson 혹은 Spearman 상관계수를 사용하여 나타낸다. 이 방법은 zero-inflated data에서 낮은 abudance를 가진 OTU 간의 가짜 상관관계 (0 값이 많은 taxa끼리 상관관계가 유사하게 나옴)를 나타낸다는 단점이 있다.
- Regression-Based Methods
- 위의 두 방법은 복잡한 다형성의 상호작용을 잡아내기 어려우며, 이는 multiple regression analysis가 대안이 될 수 있다. 그러나 overfitting의 문제가 존재하지만 이는 cross-validation으로 해결될 수 있다.
- Probabilistic Graphical Models (PGMs)
- 확률이론과 그래프 이론을 사용하여 불확실성과 복잡성을 다룬다. Bayesian networks와 Markov networks가 가장 널리 사용된다.
| network 그리는 도구들
- R 패키지: SpiecEasi, MInt, NetCoMi 등
| 미생물 네트워크 연구에서 고려해야할 점
1) 각 미생물의 상호작용이 실제로 미생물 군집의 풍부도에 영향을 미치는가?
2) 미생물 풍부도는 어떻게 처리되어야 하는가?
3) 풍부도가 낮은 taxa를 어떻게 처리하는가?
4) 환경 요인은 어떻게 처리하는가?
5) higher-order interactions (HOIs; 고차 상호작용) 은 어떠한가?
6) 미생물 네트워크를 어떻게 구축하고 평가해야 하는가?
7) 생물학적 데이터에서 미생물 네트워크 구성을 밴치마킹 하는 방법은?
8) hairball(복합하고 다루기 어려운 문제)에서 어떤 것을 배울 수 있는가?
9) 핵심 네트워크를 어떻게 알아내는가?
10) 미생물 네트워크가 생태계를 잘 반영하는가?
(Faust, K. Open challenges for microbial network construction and analysis. ISME J 15, 3111–3118 (2021). https://doi.org/10.1038/s41396-021-01027-4)
2. NetCoMi
사용할 패키지인 NetCoMi(Network Construction and Comparison for Microbiome Data)는 R 기반으로 여러 network 패키지를 통합한 프로그램이다. 이 프로그램을 통해 network plot을 그려보자.
- 논문 게재 : 2021년
- 인용 = 185
패키지에서 사용 가능한 옵션은 아래와 같다.
- 사용 가능한 네트워크 구성 방법
- Association measures: Pearson coefficient, Spearman coefficient, SparCC, SpiecEasi, CCLasso, SPRING 등
- Dissimilarity measures: Euclidean distance, Bray-Curtis dissimilarity, Aitchison distance 등
- 데이터 처리 방법
- zero replacement: pseudo count, Multiplicative replacement, Bayesian-multiplicative replacement 등
- Normalization methods: Total Sum Scaling (TSS), Cumulative Sum Scaling (CSS), Centered log-ratio (clr) transformation 등
같은 네트워크 구성 방법에서 기존 패키지와 netcomi내부에서 수행하는 결과의 차이는 나지 않았으니 안심하고 사용 가능하다.
3. NetCoMi 설치
# Required packages
install.packages("devtools")
install.packages("BiocManager")
# Install NetCoMi
devtools::install_github("stefpeschel/NetCoMi",
dependencies = c("Depends", "Imports", "LinkingTo"),
repos = c("https://cloud.r-project.org/",
BiocManager::repositories()))
devtools::install_github("zdk123/SpiecEasi")
devtools::install_github("GraceYoon/SPRING")
4. 예제 데이터
- qiime2의 예제 데이터인 moving picture 데이터를 phyloseq object로 변환하였다.
- 위 데이터에는 장, 양 손바닥과 혀의 마이크로바이옴 데이터를 담고 있다.
5. Network plot 그리기
예제 데이터의 gut 샘플만 추출하여 관찰해 보자. 스크립트는 공식 tutorial에 적혀있는 default값을 사용하였다.
- transformation: CLR
- network construction: spearman coefficient
5-1. 데이터 처리
1) 먼저 예제 데이터를 R에서 불러오고, gut 데이터만 추출한다
2) Top 100의 ASV만 추출한다
3) tax_glom을 사용하여 otu table을 Genus로 합쳐준다
4) NetCoMi::renameTaxa를 통해 Genus의 이름을 netcomi 패키지 형식에 맞게 변형한다.
library(SpiecEasi)
library(igraph)
library(NetCoMi)
library(phyloseq)
# 1.import data
ps <- readRDS("./ps.rds")
gut = subset_samples(ps, body.site %in% c("gut")) # 장 마이크로바이옴 샘플만 추출
# 2. processing data
myTaxa100 = names(sort(taxa_sums(gut), decreasing = TRUE)[1:100]) # Top 100 taxa만 추출
gut.1 = prune_taxa(myTaxa100, gut)
# 3. filtering
gut_genus <- tax_glom(gut.1, taxrank = "Genus") # Agglomerate to genus level
taxtab <- as(tax_table(gut_genus), "matrix")
# 4. 이름 수정
gut_genus_renamed <- NetCoMi::renameTaxa(gut_genus,
pat = "<name>",
substPat = "<name>_<subst_name>(<subst_R>)",
numDupli = "Genus")
5-2. 네트워크 구성
netcomi의 netConstruct 함수로 phyloseq데이터에서 네트워크를 구축할 수 있다.
이에 대한 세부 옵션을 살펴보자.
netConstruct(data, # Input 파일 data = matrix, phyloseq
data2 = NULL, # 두 번째 Input 파일 = data matrix, phyloseq
dataType = "counts", # phyloseq 넣으면 X, "counts", "correlation", "partialCorr", "condDependence", "proportionality", "dissimilarity"
taxRank = NULL,
# Association/dissimilarity measure:
measure = "spieceasi", # 네트워크 계산 방법 = "spieceasi" (default)/ [Distance] "euclidean", "bray", "kld", "jeffrey", "jsd", "ckld", "aitchison", [Correlation] "pearson", "spearman", [Others] "bicor", "sparcc", "cclasso", "ccrepe", "spring", "gcoda", "propr" as association measures, and
# Preprocessing:
filtTax = "none", # tax 필터링 = "none", "totalReads", "relFreq", "numbSamp", "highestFreq"
filtTaxPar = NULL, # 위에서 설정한 옵션에 따라 cutoff (int) 설정
filtSamp = "none", # 샘플 필터링 = "none", "totalReads", "numbTaxa", "highestFreq"
filtSampPar = NULL, # 위에서 설정한 옵션에 따라 cutoff (int) 설정
zeroMethod = "none", # 0값 처리 방법 = "none" (default), "pseudo" 모든 matrix에 1값 추가 , "pseudoZO", "multRepl", "alrEM", "bayesMult"
zeroPar = NULL,
normMethod = "none", # 데이터 테이블 표준화 방법 = "none" (default), "TSS" (or "fractions"), "CSS", "COM", "rarefy", "VST", "clr", "mclr
normPar = NULL,
# Sparsification:
sparsMethod = "t-test",# sparsification 방법, edge 구성 = "none", "t-test", "bootstrap", "threshold", "softThreshold", "knn"
thresh = 0.3, # 위 옵션에서 "threshold" 선택 시
alpha = 0.05, # "t-test"와 "bootstrap procedure" 일 때만
adjust = "adaptBH", # "t-test"와 "bootstrap procedure" 일 때만
nboot = 1000L, # number of bootstrap samples
cores = 1L, # number of CPU cores
# Transformation:
dissFunc = "signed", # associations을 dissimilarities로 바꾸는 방법 = "signed"(default), "unsigned", "signedPos", "TOMdiss".
weighted = TRUE, # similarity values are used as adjacencies.
# Further arguments:
verbose = 2, # Possible values: "0": no messages, "1": only important messages, "2"(default): all progress messages, "3" messages returned by external functions are shown in addition
seed = NULL
)
netAnalyze(net, # netConstruct output
# Centrality related:
centrLCC = TRUE, # largest connected component (LCC) 에 대한 Centrality 도 추가 계산
# Cluster related:
clustMethod = NULL, # (Defaults) "cluster_fast_greedy", "hierarchical"
clustPar = NULL, # "hierarchical" 선택했다면, "average" 등의 method 설정 가능
# Hub related:
hubPar = "eigenvector", # "degree", "betweenness", "closeness" 등도 가능, Hub란 모든 측정 방법에서 highest centrality를 가진 node를 말함
# Further arguments:
verbose = 1 # Possible values: "0": no messages, "1": only important messages, "2"(default): all progress messages are shown
)
1) netConstruct()를 사용하여 spearman 상관관계를 사용하여 네트워크를 구성할 것이다. meansure를 "spearman"으로 적고, normalization은 "clr", zero값도 "multiRepl" 방법으로 처리해 주자.
2) netAnalyze()로 구성된 네트워크의 cluster를 측정하자. 이는 "cluster_fast_greedy" 알고리즘을 사용할 것이다.
# 4. 네크워크 구성
# Network construction and analysis
net_genus <- netConstruct(gut_genus_renamed, # phyloseq
taxRank = "Genus", # 기준 tax level
measure = "spearman", # spearman correlation 으로 계산해보자
zeroMethod = "multRepl",
normMethod = "clr", # transformation
sparsMethod = "threshold",
thresh = 0.3,
verbose = 3)
props_genus <- netAnalyze(net_genus, clustMethod = "cluster_fast_greedy")
plotHeat(mat = props_genus$graphletLCC$gcm1,
pmat = props_genus$graphletLCC$pAdjust1,
type = "mixed",
title = "GCM",
colorLim = c(-1, 1),
mar = c(2, 0, 2, 0))
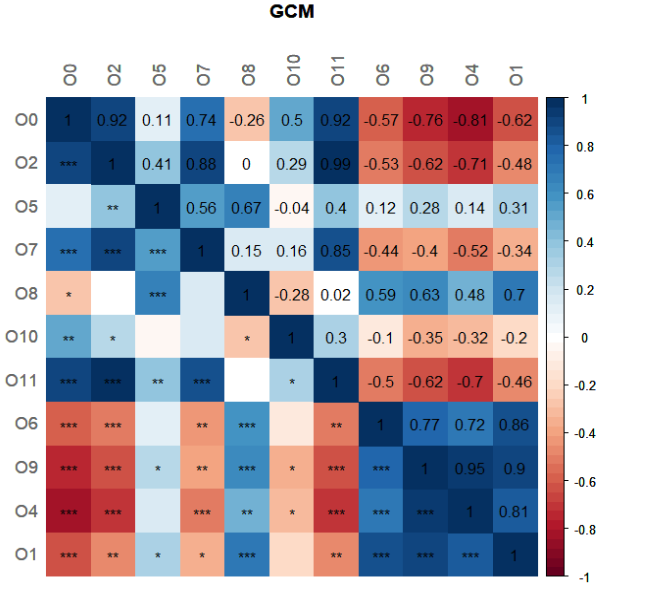
3) 만들어진 네트워크의 정보를 살펴보자
summary(props_genus, numbNodes = 5L)
# Component sizes
#
# size: 36 <- Node 개수
# #: 1 <- Singletone 개수
# ______________________________
# Global network properties
# ``````````````````````````````````
# Whole network:
#
# Number of components 1.00000
# Clustering coefficient 0.69085
# Modularity 0.02265
# Positive edge percentage 44.10256
# Edge density 0.61905 <- density와 connectivity가 낮을 수록 네트워크의 견고함이 낮음
# Natural connectivity 0.16586
# Vertex connectivity 11.00000
# Edge connectivity 11.00000
# Average dissimilarity* 0.80056
# Average path length** 0.98561 <- 서로 다른 node로 가기 위해 0.98 step이 필요
# -----
# *: Dissimilarity = 1 - edge weight
# **: Path length = Units with average dissimilarity
#
#
# ______________________________
# Clusters
# - In the whole network
# - Algorithm: cluster_fast_greedy
# ``````````````````````````````````
#
# name: 1 2 3 Cluster 개수
# #: 8 16 12 각 Cluster의 node 개수
#
# ______________________________
# Hubs
# - In alphabetical/numerical order
# - Based on empirical quantiles of centralities
# `````````````````````````````````` <- 총 두 개의 허브 노드 계산
# Odoribacter
# Parabacteroides
#
# ______________________________
# Centrality measures
# - In decreasing order
# - Computed for the complete network
# ``````````````````````````````````
# Degree (normalized):
#
# Odoribacter 0.82857
# Parabacteroides 0.82857
# Gemmiger 0.80000
# Butyricimonas 0.80000
# Unclassified Bacteroidales 0.80000
#
# Betweenness centrality (normalized):
#
# Unclassified Lachnospiraceae 0.03193
# Blautia 0.02773
# Phascolarctobacterium 0.02689
# Clostridium2 0.02521
# Dorea 0.02353
#
# Closeness centrality (normalized):
#
# Odoribacter 2.01091
# Butyricimonas 1.87396
# Parabacteroides 1.85956
# Alistipes 1.65083
# Gemmiger 1.64085
#
# Eigenvector centrality (normalized):
#
# Parabacteroides 1.00000
# Odoribacter 0.98163
# Unclassified Bacteroidales 0.97918
# Gemmiger 0.97918
# Alistipes 0.96318
위 데이터로 알 수 있는 것은 아래와 같다.
- 전체 node는 36개, Singletone의 개수는 1개
- Hub node는 Odoribacter, Parabacteroides 총 두 개
- Cluster의 수는 총 3개로 각 8, 16, 12 개의 node를 포함한다
- Connectivity가 매우 높음 ->네트워크의 견고함을 반영
5-3. 레이아웃 설정
- igraph를 사용하여 네트워크의 레이아웃을 설정해준다.
- 위 단계를 건너뛰고 netcomi에 기본으로 있는 "spring" 방법을 사용해도 된다.
# 5. plot layout 설정하기
graph3 <- igraph::graph_from_adjacency_matrix(net_genus$adjaMat1,
weighted = TRUE)
set.seed(42) # 모양 고정
lay_fr <- igraph::layout_with_fr(graph3)
rownames(lay_fr) <- rownames(net_genus$adjaMat1) # Row names of the layout matrix must match the node names
5-4. network plot 그리기
# 6. network plot 그리기
plot(props_genus, # Network 구성성분
layout = lay_fr, # Network plot 의 layout # "spring" 도 사용가능
labelLength = 10, # 라벨의 길이
cexNodes = 0.8, # node의 크기
cexHubs = 1.1, # node 중 중심이 되는 node의 크기
colorVec = phylcol, # edge의 색
posCol = "darkturquoise", # positive correlation edge의 색
negCol = "orange", # negative correlation edge의 색
cexLabels = 2, # node의 라벨 크기
title1 = "Network on genus level with Spearman correlations", # 제목
showTitle = TRUE, # 제목 보이기
cexTitle = 2.3) # 제목 크기
# Get phyla names
taxtab <- as(tax_table(gut_genus_renamed), "matrix")
phyla <- as.factor(gsub("p__", "", taxtab[, "Phylum"]))
names(phyla) <- taxtab[, "Phylum"]
# index color
phylcol <- c("cyan", "blue3", "red", "lawngreen", "yellow", "deeppink")
phylcol_transp <- colToTransp(phylcol, 60)
# Phylum color index
legend(-1.1, 1.1, cex = 1, pt.cex = 1, title = "Phylum:",
legend=levels(phyla), col = phylcol_transp, bty = "n", pch = 16)
# correlation color index
legend(0.4, 1.1, cex = 1, title = "estimated correlation:",
legend = c("+","-"), lty = 1, lwd = 3, col = c("darkturquoise","orange"),
bty = "n", horiz = TRUE)
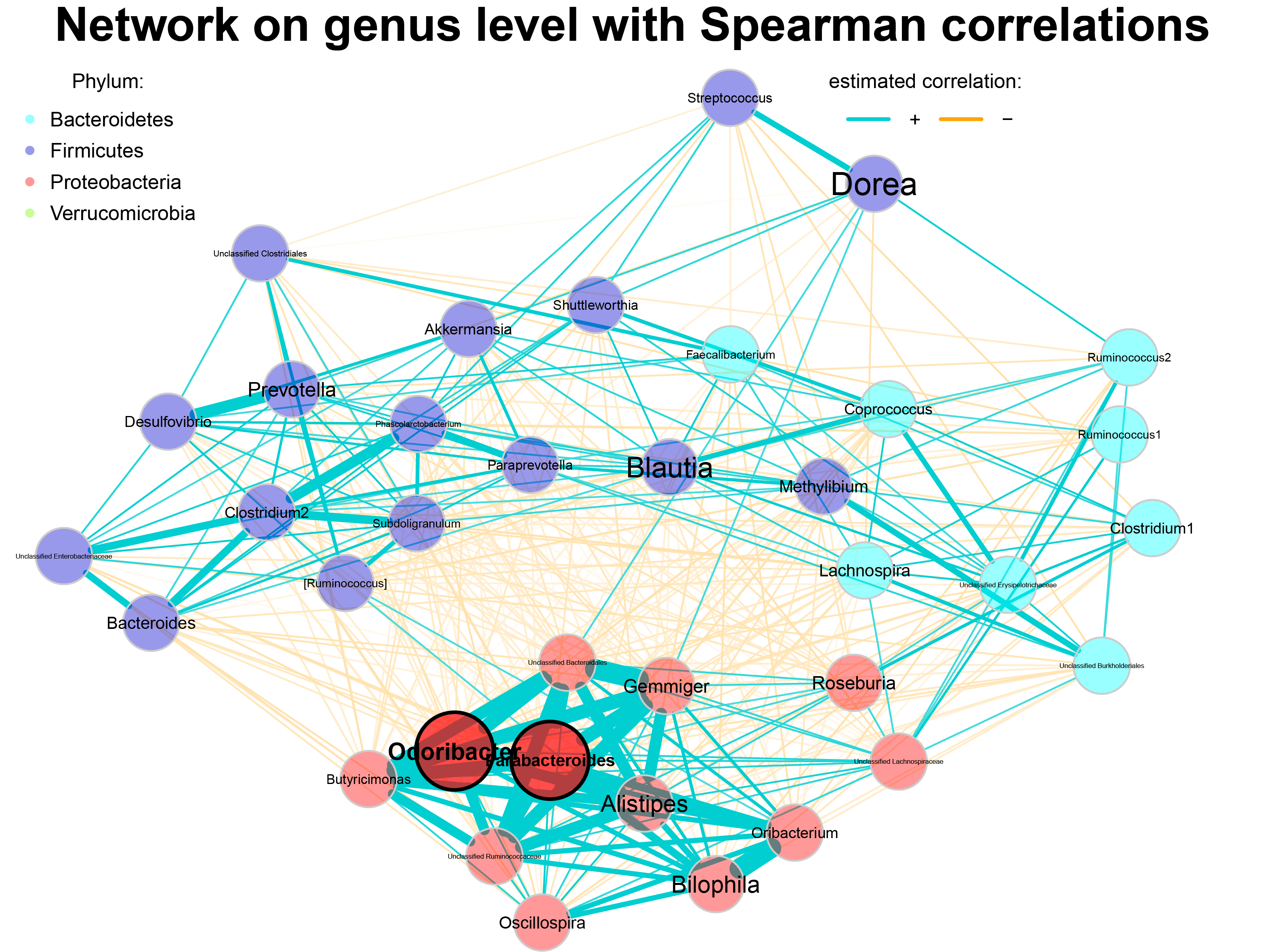
- Phylum 에 따라서 Node의 색을 지정하였으며, 선은 상관관계에 따라 색을 달리 하였다.
6. 두 그룹에서 Network plot 비교하기
6-1. 데이터 처리
이번에는 왼손과 혀의 마이크로바이옴 네트워크를 비교해 보자.
- transformation: CLR
- network construction: sparCC
library(phyloseq)
# import data
ps <- readRDS("./ps.rds")
ps.pt <- ps %>% subset_samples(body.site %in% c("left palm", "tongue")) # 770 taxa and 17 samples
# zero taxa 제거
ps.f = prune_samples(sample_sums(ps.pt)>0, ps.pt)
ps.f = prune_taxa(rowSums(otu_table(ps.f)) > 0, ps.f) # 431 taxa and 17 samples
# Agglomerate to genus level
ps_genus <- tax_glom(ps.f, taxrank = "Genus")
# Taxonomic table
taxtab <- as(tax_table(ps_genus), "matrix")6-2. 네트워크 구성
- NetComi 형식으로 변환
# necomi 형식으로 변환 및 Genus level 설정
ps_genus_renamed <- NetCoMi::renameTaxa(ps_genus,
pat = "<name>",
substPat = "<name>_<subst_name>(<subst_R>)",
numDupli = "Genus")
# 각 부위에 대한 샘플 추출
ps_genus_renamed_palm <- phyloseq::subset_samples(ps_genus_renamed,
body.site == "left palm")
ps_genus_renamed_tongue <- phyloseq::subset_samples(ps_genus_renamed,
body.site == "tongue")
- 네트워크 구성
# Network construction and analysis
ps_genus.sparcc <- netConstruct(
data = ps_genus_renamed_palm,
data2 = ps_genus_renamed_tongue,
filtTax = "highestFreq", # Top을 뽑는 기준
filtTaxPar = list(highestFreq = 100), # Top 100
taxRank = "Genus", # Genus level
measure = "sparcc", # sparcc
measurePar = list(nlambda=20, rep.num=10),
normMethod = "clr", # transformation
zeroMethod = "none", # zero 값 보정
sparsMethod = "threshold",
adjust = "adaptBH", # p-value 값 보정
thresh = 0.3,
dissFunc = "signed",
verbose = 2,
seed = 42)
- Hub노드 구성 및 결과 보기
ps_props_sparcc <- netAnalyze(ps_genus.sparcc,
clustMethod = "cluster_fast_greedy",
hubPar = "eigenvector", # Hub node 판별방법
normDeg = FALSE)
summary(ps_props_sparcc, groupNames = c("left palm", "tongue"))
#
# Component sizes <- node개수
# # ```````````````
# left palm:
# size: 61
# #: 1
# tongue:
# size: 33 1
# #: 1 28
# ______________________________
# Global network properties <- 네트워크의 연결성, 견고함 등을 나타냄
# `````````````````````````
# Largest connected component (LCC):
# left palm tongue
# Relative LCC size 1.00000 0.54098
# Clustering coefficient 0.65197 0.57913
# Modularity 0.12369 0.12694
# Positive edge percentage 49.54338 59.89583
# Edge density 0.47869 0.36364
# Natural connectivity 0.12729 0.10811
# Vertex connectivity 10.00000 2.00000
# Edge connectivity 10.00000 2.00000
# Average dissimilarity* 0.84269 0.86923
# Average path length** 1.04898 1.13962
#
# Whole network:
# left palm tongue
# Number of components 1.00000 29.00000
# Clustering coefficient 0.65197 0.57913
# Modularity 0.12369 0.12694
# Positive edge percentage 49.54338 59.89583
# Edge density 0.47869 0.10492
# Natural connectivity 0.12729 0.04686
# -----
# *: Dissimilarity = 1 - edge weight
# **: Path length = Units with average dissimilarity
#
# ______________________________
# Clusters <- 각 네트워크의 cluster 수와 그 안의 노드 개수
# - In the whole network
# - Algorithm: cluster_fast_greedy
# ````````````````````````````````
# left palm:
# name: 1 2 3
# #: 19 23 19
#
# tongue:
# name: 0 1 2 3
# #: 28 10 13 10
#
# ______________________________
# Hubs <- 허브 노드(아래 네가지 방법에서 공통적인 taxa)
# - In alphabetical/numerical order
# - Based on empirical quantiles of centralities
# ```````````````````````````````````````````````
# left palm tongue
# Fusobacterium Campylobacter
# Leptotrichia Leptotrichia
# Oribacterium Unclassified Lachnospiraceae
#
# ______________________________
# Centrality measures
# - In decreasing order
# - Centrality of disconnected components is zero
# ````````````````````````````````````````````````
# Degree (unnormalized):
# left palm tongue
# Unclassified Streptophyta 47 10
# Leptotrichia 47 20
# Bacteroides 46 10
# Unclassified Bacteria 46 0
# Fusobacterium 45 15
# ______ ______
# Leptotrichia 47 20
# Campylobacter 36 20
# Oribacterium 39 20
# Haemophilus 24 18
# [Prevotella] 10 18
#
# Betweenness centrality (normalized):
# left palm tongue
# Fusobacterium 0.05085 0.02419
# Leptotrichia 0.04181 0.0504
# Bacteroides 0.02881 0.01008
# Unclassified Streptophyta 0.02825 0.00403
# Parvimonas 0.02825 0
# ______ ______
# Oribacterium 0.02316 0.14516
# Haemophilus 0.00226 0.08468
# [Prevotella] 0.00282 0.05645
# Rothia 0.00734 0.05444
# Leptotrichia 0.04181 0.0504
#
# Closeness centrality (normalized):
# left palm tongue
# Fusobacterium 1.34753 1.20688
# Neisseria 1.3215 1.14746
# Haemophilus 1.31325 1.21906
# Unclassified Bacteria 1.28898 0
# Leptotrichia 1.2852 1.33367
# ______ ______
# [Prevotella] 0.82772 1.34515
# Leptotrichia 1.2852 1.33367
# Unclassified Lachnospiraceae 1.10444 1.28345
# Campylobacter 1.15364 1.27294
# Veillonella 1.11835 1.24568
#
# Eigenvector centrality (normalized):
# left palm tongue
# Fusobacterium 1 0.84707
# Leptotrichia 0.99148 1
# Oribacterium 0.8844 0.79039
# Unclassified Bacteria 0.86111 0
# Unclassified Streptophyta 0.8609 0.21985
# ______ ______
# Leptotrichia 0.99148 1
# Unclassified Lachnospiraceae 0.61801 0.91662
# Campylobacter 0.80563 0.89528
# Fusobacterium 1 0.84707
# [Prevotella] 0.13195 0.83545
- 왼손바닥의 node수는node 수는 61, 혀에서 single tone을 제외한 node 수는 33개로 구성된다
- 왼손바닥은 3개의 cluster를 가지며, 혀는 4개의 cluster로 구성된다.
- 왼손바닥은 혀보다 connectivity가 높아 더 견고한 네트워크임을 알 수 있다. 견고한 네트워크는 외부의 영향에도 안정적인 네트워크를 더 잘 구성한다고 해석할 수 있음
- 왼손바닥의 허브노드는 Fusobacterium,Leptotrichia, Oribacterium Unclassified이며, 혀의 허브노드는 Campylobacter, Leptotrichia, Lachnospiraceae이다.
6-3. 레이아웃 설정
# Compute layout
ps_graph <- igraph::graph_from_adjacency_matrix(ps_genus.sparcc$adjaMat1,
weighted = TRUE)
set.seed(42)
ps_lay_fr <- igraph::layout_with_fr(ps_graph)
# Row names of the layout matrix must match the node names
rownames(ps_lay_fr) <- rownames(ps_genus.sparcc$adjaMat1)
6-4. network plot 그리기
# Get phyla names
taxtab <- as(tax_table(ps_genus_renamed), "matrix")
phyla <- as.factor(taxtab[, "Phylum"])
names(phyla) <- taxtab[, "Genus"]
table(phyla)
# Define phylum colors
library(RColorBrewer)
phylcol <- c(brewer.pal(12, "Paired"), brewer.pal(12, "Set3"))
# Colors used in the legend should be equally transparent as in the plot
phylcol_transp <- colToTransp(phylcol, 60)
plot(ps_props_sparcc,
layout = "spring",
nodeSize = "eigenvector", # "clr",
nodeColor = "cluster", #"feature",
rmSingles = T,
cexNodes = 0.8,
cexHubs = 1.1,
featVecCol = phyla,
colorVec = phylcol,
posCol = "darkturquoise",
negCol = "orange",
title1 = "Network on genus level with sparcc correlations",
showTitle = TRUE,
groupNames = c("left palm","tongue"))
legend(-1.2, 10, cex = 1.5, pt.cex = 2.5, title = "Phylum:",
legend=levels(phyla), col = phylcol_transp, bty = "n", pch = 16)
legend(0.7, 1.0, cex = 2.2, title = "estimated correlation:",
legend = c("+","-"), lty = 1, lwd = 3, col = c("darkturquoise","orange"),
bty = "n", horiz = TRUE)
dev.off()
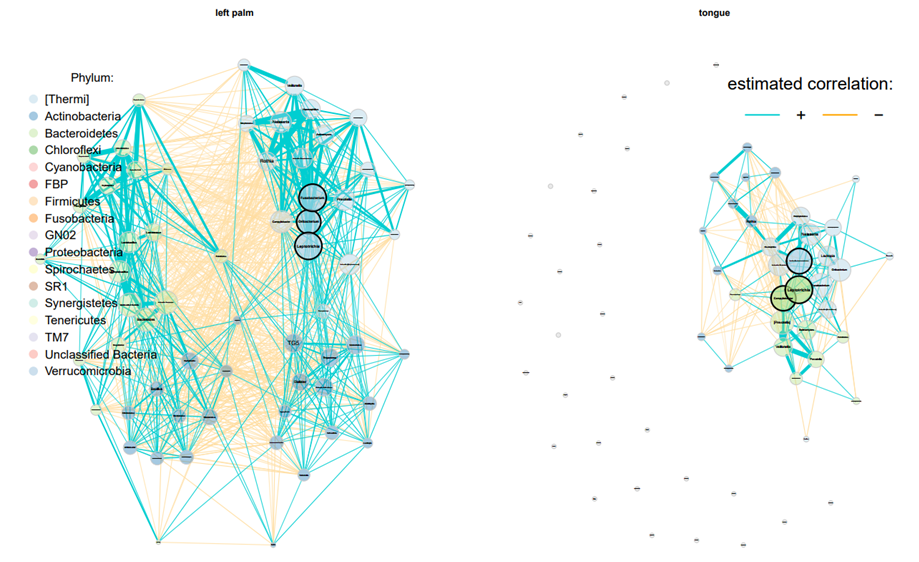
- 위 그림도 각각의 node색을 phylum에 따라 지정하였으며, node의 크기는 eigenvector에 따라 달라진다.
- 이전 네트워크 그림과 다르게 igraph로 만든 레이아웃이 아닌 "spring"을 사용하였다.
네트워크 분석 결과, 혀의 마이크로바이옴에 비해 왼손바닥의 마이크로바이옴이 더 다양하고 더 많은 cluster로 구성되며, Genus level에서 각 node에 대한 연결이 견고하다. 왼손바닥의 중심이 되는 genus는 Fusobacterium, Leptotrichia, Oribacterium Unclassified이며, 혀는 Campylobacter, Leptotrichia, Lachnospiraceae임을 알 수 있다.
기타

- 올해 나온 ggCluster는 NetCoMi보다 구동 방법이 쉬워 보이지만, 아직 사용해보지는 않았다. 더 쉬운 패키지를 찾는 다면 추천한다.
- tutorial (중국어)
https://blog.csdn.net/woodcorpse/article/details/125863402
https://blog.csdn.net/weixin_60157921/article/details/129548610
https://zhuanlan.zhihu.com/p/617569543
Reference
- Matloff, N, A User’s Guide to Network Analysis in R. Journal of Statistical Software, Book Reviews, 72(3), 2016, 1–2. https://doi.org/10.18637/jss.v072.b03
- Layeghifard et al., Disentangling Interactions in the Microbiome: A Network Perspectiv. Trends in Microbiology, 2017
- The Genetic Diversity Centre (GDC), Microbiota Data Analysis 2020 Workshop- Microbial Networks, 2020 (https://www.gdc-docs.ethz.ch/MDA/handouts/MDA20_Network_Analysis.pdf)
- https://microbiome.github.io/OMA/network-learning.html#network-analysis-with-netcomi
- Stefanie Peschel, Christian L Müller, Erika von Mutius, Anne-Laure Boulesteix, Martin Depner, NetCoMi: network construction and comparison for microbiome data in R, Briefings in Bioinformatics, Volume 22, Issue 4, July 2021, bbaa290, https://doi.org/10.1093/bib/bbaa290
글 작성: 24.01.03.
내용추가: 24.01.02.
1. Network analysis
| 마이크로바이옴 연구에서 네트워크 연구란?
- microbiome은 복잡한 미생물 군집을 말한다. 여러 생물들의 상호작용은 전체 미생물의 구조를 안정적이며 견고하게 만든다.
- 네트워크 plot은 미생물의 scale과 다양성을 모두 나타낼 수 있으며, 단편적인 관계보다 보편적인 시스템을 보여준다.
| Network의 구성성분
- node: 하나의 샘플이나 OTU/ASV를 뜻한다
- edge: 각 단위 간의 관계를 말한다
| network plot에서 알 수 있는 정보들
- node의 정보: 색, 모양, 크기, 라벨에 정보를 표시할 수 있다.
- e.g. 색에는 그룹 데이터를, 크기는 relative abundance를 나타낼 수 있다
- Edge 정보 : dge의 두께, 색, 모양에 따라 정보를 표시할 수 있다.
- e.g. 빨간색은 음의 상관계수, 초록색은 양의 상관계수를 나타냄
- 군집의 정보: 독립적인 생물군과 cluster를 구분 지을 수 있다
- Network Dynamic: 시간에 따라 네트워크 간의 전체 적인 연결 변화와 abundance변화를 관찰할 수 있다
- Hub(Keystones) 정보: 전체 네트워크에서 상호작용이 가장 많은 key stone 생물을 찾을 수 있다. 이는 각 node의 Centrality를 측정하며, 측정 방법은 Degree, Closeness, Betweenness 등이 있다.
Betweenness centrality
| network 알고리즘
- Dissimilarity-Based Methods
- 가장 간단하고 빠른 방법으로, pairwise dissimilarity index(e.g. Bray-Cutris) 등을 이용하는 방법이다.
- phyloseq 패키지는 위 방법을 지원하고 있다.
- Correlation-Based Methods
- 상관관계 기반 방법은 Dissimilarity-Based의 대안으로 가장 인기 있는 방법이다. OTU 간의 pairwise 상호관계를 Pearson 혹은 Spearman 상관계수를 사용하여 나타낸다. 이 방법은 zero-inflated data에서 낮은 abudance를 가진 OTU 간의 가짜 상관관계 (0 값이 많은 taxa끼리 상관관계가 유사하게 나옴)를 나타낸다는 단점이 있다.
- Regression-Based Methods
- 위의 두 방법은 복잡한 다형성의 상호작용을 잡아내기 어려우며, 이는 multiple regression analysis가 대안이 될 수 있다. 그러나 overfitting의 문제가 존재하지만 이는 cross-validation으로 해결될 수 있다.
- Probabilistic Graphical Models (PGMs)
- 확률이론과 그래프 이론을 사용하여 불확실성과 복잡성을 다룬다. Bayesian networks와 Markov networks가 가장 널리 사용된다.
| network 그리는 도구들
- R 패키지: SpiecEasi, MInt, NetCoMi 등
| 미생물 네트워크 연구에서 고려해야할 점
1) 각 미생물의 상호작용이 실제로 미생물 군집의 풍부도에 영향을 미치는가?
2) 미생물 풍부도는 어떻게 처리되어야 하는가?
3) 풍부도가 낮은 taxa를 어떻게 처리하는가?
4) 환경 요인은 어떻게 처리하는가?
5) higher-order interactions (HOIs; 고차 상호작용) 은 어떠한가?
6) 미생물 네트워크를 어떻게 구축하고 평가해야 하는가?
7) 생물학적 데이터에서 미생물 네트워크 구성을 밴치마킹 하는 방법은?
8) hairball(복합하고 다루기 어려운 문제)에서 어떤 것을 배울 수 있는가?
9) 핵심 네트워크를 어떻게 알아내는가?
10) 미생물 네트워크가 생태계를 잘 반영하는가?
(Faust, K. Open challenges for microbial network construction and analysis. ISME J 15, 3111–3118 (2021). https://doi.org/10.1038/s41396-021-01027-4)
2. NetCoMi
사용할 패키지인 NetCoMi(Network Construction and Comparison for Microbiome Data)는 R 기반으로 여러 network 패키지를 통합한 프로그램이다. 이 프로그램을 통해 network plot을 그려보자.
- 논문 게재 : 2021년
- 인용 = 185
패키지에서 사용 가능한 옵션은 아래와 같다.
- 사용 가능한 네트워크 구성 방법
- Association measures: Pearson coefficient, Spearman coefficient, SparCC, SpiecEasi, CCLasso, SPRING 등
- Dissimilarity measures: Euclidean distance, Bray-Curtis dissimilarity, Aitchison distance 등
- 데이터 처리 방법
- zero replacement: pseudo count, Multiplicative replacement, Bayesian-multiplicative replacement 등
- Normalization methods: Total Sum Scaling (TSS), Cumulative Sum Scaling (CSS), Centered log-ratio (clr) transformation 등
같은 네트워크 구성 방법에서 기존 패키지와 netcomi내부에서 수행하는 결과의 차이는 나지 않았으니 안심하고 사용 가능하다.
3. NetCoMi 설치
# Required packages
install.packages("devtools")
install.packages("BiocManager")
# Install NetCoMi
devtools::install_github("stefpeschel/NetCoMi",
dependencies = c("Depends", "Imports", "LinkingTo"),
repos = c("https://cloud.r-project.org/",
BiocManager::repositories()))
devtools::install_github("zdk123/SpiecEasi")
devtools::install_github("GraceYoon/SPRING")
4. 예제 데이터
- qiime2의 예제 데이터인 moving picture 데이터를 phyloseq object로 변환하였다.
- 위 데이터에는 장, 양 손바닥과 혀의 마이크로바이옴 데이터를 담고 있다.
5. Network plot 그리기
예제 데이터의 gut 샘플만 추출하여 관찰해 보자. 스크립트는 공식 tutorial에 적혀있는 default값을 사용하였다.
- transformation: CLR
- network construction: spearman coefficient
5-1. 데이터 처리
1) 먼저 예제 데이터를 R에서 불러오고, gut 데이터만 추출한다
2) Top 100의 ASV만 추출한다
3) tax_glom을 사용하여 otu table을 Genus로 합쳐준다
4) NetCoMi::renameTaxa를 통해 Genus의 이름을 netcomi 패키지 형식에 맞게 변형한다.
library(SpiecEasi)
library(igraph)
library(NetCoMi)
library(phyloseq)
# 1.import data
ps <- readRDS("./ps.rds")
gut = subset_samples(ps, body.site %in% c("gut")) # 장 마이크로바이옴 샘플만 추출
# 2. processing data
myTaxa100 = names(sort(taxa_sums(gut), decreasing = TRUE)[1:100]) # Top 100 taxa만 추출
gut.1 = prune_taxa(myTaxa100, gut)
# 3. filtering
gut_genus <- tax_glom(gut.1, taxrank = "Genus") # Agglomerate to genus level
taxtab <- as(tax_table(gut_genus), "matrix")
# 4. 이름 수정
gut_genus_renamed <- NetCoMi::renameTaxa(gut_genus,
pat = "<name>",
substPat = "<name>_<subst_name>(<subst_R>)",
numDupli = "Genus")
5-2. 네트워크 구성
netcomi의 netConstruct 함수로 phyloseq데이터에서 네트워크를 구축할 수 있다.
이에 대한 세부 옵션을 살펴보자.
netConstruct(data, # Input 파일 data = matrix, phyloseq
data2 = NULL, # 두 번째 Input 파일 = data matrix, phyloseq
dataType = "counts", # phyloseq 넣으면 X, "counts", "correlation", "partialCorr", "condDependence", "proportionality", "dissimilarity"
taxRank = NULL,
# Association/dissimilarity measure:
measure = "spieceasi", # 네트워크 계산 방법 = "spieceasi" (default)/ [Distance] "euclidean", "bray", "kld", "jeffrey", "jsd", "ckld", "aitchison", [Correlation] "pearson", "spearman", [Others] "bicor", "sparcc", "cclasso", "ccrepe", "spring", "gcoda", "propr" as association measures, and
# Preprocessing:
filtTax = "none", # tax 필터링 = "none", "totalReads", "relFreq", "numbSamp", "highestFreq"
filtTaxPar = NULL, # 위에서 설정한 옵션에 따라 cutoff (int) 설정
filtSamp = "none", # 샘플 필터링 = "none", "totalReads", "numbTaxa", "highestFreq"
filtSampPar = NULL, # 위에서 설정한 옵션에 따라 cutoff (int) 설정
zeroMethod = "none", # 0값 처리 방법 = "none" (default), "pseudo" 모든 matrix에 1값 추가 , "pseudoZO", "multRepl", "alrEM", "bayesMult"
zeroPar = NULL,
normMethod = "none", # 데이터 테이블 표준화 방법 = "none" (default), "TSS" (or "fractions"), "CSS", "COM", "rarefy", "VST", "clr", "mclr
normPar = NULL,
# Sparsification:
sparsMethod = "t-test",# sparsification 방법, edge 구성 = "none", "t-test", "bootstrap", "threshold", "softThreshold", "knn"
thresh = 0.3, # 위 옵션에서 "threshold" 선택 시
alpha = 0.05, # "t-test"와 "bootstrap procedure" 일 때만
adjust = "adaptBH", # "t-test"와 "bootstrap procedure" 일 때만
nboot = 1000L, # number of bootstrap samples
cores = 1L, # number of CPU cores
# Transformation:
dissFunc = "signed", # associations을 dissimilarities로 바꾸는 방법 = "signed"(default), "unsigned", "signedPos", "TOMdiss".
weighted = TRUE, # similarity values are used as adjacencies.
# Further arguments:
verbose = 2, # Possible values: "0": no messages, "1": only important messages, "2"(default): all progress messages, "3" messages returned by external functions are shown in addition
seed = NULL
)
netAnalyze(net, # netConstruct output
# Centrality related:
centrLCC = TRUE, # largest connected component (LCC) 에 대한 Centrality 도 추가 계산
# Cluster related:
clustMethod = NULL, # (Defaults) "cluster_fast_greedy", "hierarchical"
clustPar = NULL, # "hierarchical" 선택했다면, "average" 등의 method 설정 가능
# Hub related:
hubPar = "eigenvector", # "degree", "betweenness", "closeness" 등도 가능, Hub란 모든 측정 방법에서 highest centrality를 가진 node를 말함
# Further arguments:
verbose = 1 # Possible values: "0": no messages, "1": only important messages, "2"(default): all progress messages are shown
)
1) netConstruct()를 사용하여 spearman 상관관계를 사용하여 네트워크를 구성할 것이다. meansure를 "spearman"으로 적고, normalization은 "clr", zero값도 "multiRepl" 방법으로 처리해 주자.
2) netAnalyze()로 구성된 네트워크의 cluster를 측정하자. 이는 "cluster_fast_greedy" 알고리즘을 사용할 것이다.
# 4. 네크워크 구성
# Network construction and analysis
net_genus <- netConstruct(gut_genus_renamed, # phyloseq
taxRank = "Genus", # 기준 tax level
measure = "spearman", # spearman correlation 으로 계산해보자
zeroMethod = "multRepl",
normMethod = "clr", # transformation
sparsMethod = "threshold",
thresh = 0.3,
verbose = 3)
props_genus <- netAnalyze(net_genus, clustMethod = "cluster_fast_greedy")
plotHeat(mat = props_genus$graphletLCC$gcm1,
pmat = props_genus$graphletLCC$pAdjust1,
type = "mixed",
title = "GCM",
colorLim = c(-1, 1),
mar = c(2, 0, 2, 0))
3) 만들어진 네트워크의 정보를 살펴보자
summary(props_genus, numbNodes = 5L)
# Component sizes
#
# size: 36 <- Node 개수
# #: 1 <- Singletone 개수
# ______________________________
# Global network properties
# ``````````````````````````````````
# Whole network:
#
# Number of components 1.00000
# Clustering coefficient 0.69085
# Modularity 0.02265
# Positive edge percentage 44.10256
# Edge density 0.61905 <- density와 connectivity가 낮을 수록 네트워크의 견고함이 낮음
# Natural connectivity 0.16586
# Vertex connectivity 11.00000
# Edge connectivity 11.00000
# Average dissimilarity* 0.80056
# Average path length** 0.98561 <- 서로 다른 node로 가기 위해 0.98 step이 필요
# -----
# *: Dissimilarity = 1 - edge weight
# **: Path length = Units with average dissimilarity
#
#
# ______________________________
# Clusters
# - In the whole network
# - Algorithm: cluster_fast_greedy
# ``````````````````````````````````
#
# name: 1 2 3 Cluster 개수
# #: 8 16 12 각 Cluster의 node 개수
#
# ______________________________
# Hubs
# - In alphabetical/numerical order
# - Based on empirical quantiles of centralities
# `````````````````````````````````` <- 총 두 개의 허브 노드 계산
# Odoribacter
# Parabacteroides
#
# ______________________________
# Centrality measures
# - In decreasing order
# - Computed for the complete network
# ``````````````````````````````````
# Degree (normalized):
#
# Odoribacter 0.82857
# Parabacteroides 0.82857
# Gemmiger 0.80000
# Butyricimonas 0.80000
# Unclassified Bacteroidales 0.80000
#
# Betweenness centrality (normalized):
#
# Unclassified Lachnospiraceae 0.03193
# Blautia 0.02773
# Phascolarctobacterium 0.02689
# Clostridium2 0.02521
# Dorea 0.02353
#
# Closeness centrality (normalized):
#
# Odoribacter 2.01091
# Butyricimonas 1.87396
# Parabacteroides 1.85956
# Alistipes 1.65083
# Gemmiger 1.64085
#
# Eigenvector centrality (normalized):
#
# Parabacteroides 1.00000
# Odoribacter 0.98163
# Unclassified Bacteroidales 0.97918
# Gemmiger 0.97918
# Alistipes 0.96318
위 데이터로 알 수 있는 것은 아래와 같다.
- 전체 node는 36개, Singletone의 개수는 1개
- Hub node는 Odoribacter, Parabacteroides 총 두 개
- Cluster의 수는 총 3개로 각 8, 16, 12 개의 node를 포함한다
- Connectivity가 매우 높음 ->네트워크의 견고함을 반영
5-3. 레이아웃 설정
- igraph를 사용하여 네트워크의 레이아웃을 설정해준다.
- 위 단계를 건너뛰고 netcomi에 기본으로 있는 "spring" 방법을 사용해도 된다.
# 5. plot layout 설정하기
graph3 <- igraph::graph_from_adjacency_matrix(net_genus$adjaMat1,
weighted = TRUE)
set.seed(42) # 모양 고정
lay_fr <- igraph::layout_with_fr(graph3)
rownames(lay_fr) <- rownames(net_genus$adjaMat1) # Row names of the layout matrix must match the node names
5-4. network plot 그리기
# 6. network plot 그리기
plot(props_genus, # Network 구성성분
layout = lay_fr, # Network plot 의 layout # "spring" 도 사용가능
labelLength = 10, # 라벨의 길이
cexNodes = 0.8, # node의 크기
cexHubs = 1.1, # node 중 중심이 되는 node의 크기
colorVec = phylcol, # edge의 색
posCol = "darkturquoise", # positive correlation edge의 색
negCol = "orange", # negative correlation edge의 색
cexLabels = 2, # node의 라벨 크기
title1 = "Network on genus level with Spearman correlations", # 제목
showTitle = TRUE, # 제목 보이기
cexTitle = 2.3) # 제목 크기
# Get phyla names
taxtab <- as(tax_table(gut_genus_renamed), "matrix")
phyla <- as.factor(gsub("p__", "", taxtab[, "Phylum"]))
names(phyla) <- taxtab[, "Phylum"]
# index color
phylcol <- c("cyan", "blue3", "red", "lawngreen", "yellow", "deeppink")
phylcol_transp <- colToTransp(phylcol, 60)
# Phylum color index
legend(-1.1, 1.1, cex = 1, pt.cex = 1, title = "Phylum:",
legend=levels(phyla), col = phylcol_transp, bty = "n", pch = 16)
# correlation color index
legend(0.4, 1.1, cex = 1, title = "estimated correlation:",
legend = c("+","-"), lty = 1, lwd = 3, col = c("darkturquoise","orange"),
bty = "n", horiz = TRUE)
- Phylum 에 따라서 Node의 색을 지정하였으며, 선은 상관관계에 따라 색을 달리 하였다.
6. 두 그룹에서 Network plot 비교하기
6-1. 데이터 처리
이번에는 왼손과 혀의 마이크로바이옴 네트워크를 비교해 보자.
- transformation: CLR
- network construction: sparCC
library(phyloseq)
# import data
ps <- readRDS("./ps.rds")
ps.pt <- ps %>% subset_samples(body.site %in% c("left palm", "tongue")) # 770 taxa and 17 samples
# zero taxa 제거
ps.f = prune_samples(sample_sums(ps.pt)>0, ps.pt)
ps.f = prune_taxa(rowSums(otu_table(ps.f)) > 0, ps.f) # 431 taxa and 17 samples
# Agglomerate to genus level
ps_genus <- tax_glom(ps.f, taxrank = "Genus")
# Taxonomic table
taxtab <- as(tax_table(ps_genus), "matrix")6-2. 네트워크 구성
- NetComi 형식으로 변환
# necomi 형식으로 변환 및 Genus level 설정
ps_genus_renamed <- NetCoMi::renameTaxa(ps_genus,
pat = "<name>",
substPat = "<name>_<subst_name>(<subst_R>)",
numDupli = "Genus")
# 각 부위에 대한 샘플 추출
ps_genus_renamed_palm <- phyloseq::subset_samples(ps_genus_renamed,
body.site == "left palm")
ps_genus_renamed_tongue <- phyloseq::subset_samples(ps_genus_renamed,
body.site == "tongue")
- 네트워크 구성
# Network construction and analysis
ps_genus.sparcc <- netConstruct(
data = ps_genus_renamed_palm,
data2 = ps_genus_renamed_tongue,
filtTax = "highestFreq", # Top을 뽑는 기준
filtTaxPar = list(highestFreq = 100), # Top 100
taxRank = "Genus", # Genus level
measure = "sparcc", # sparcc
measurePar = list(nlambda=20, rep.num=10),
normMethod = "clr", # transformation
zeroMethod = "none", # zero 값 보정
sparsMethod = "threshold",
adjust = "adaptBH", # p-value 값 보정
thresh = 0.3,
dissFunc = "signed",
verbose = 2,
seed = 42)
- Hub노드 구성 및 결과 보기
ps_props_sparcc <- netAnalyze(ps_genus.sparcc,
clustMethod = "cluster_fast_greedy",
hubPar = "eigenvector", # Hub node 판별방법
normDeg = FALSE)
summary(ps_props_sparcc, groupNames = c("left palm", "tongue"))
#
# Component sizes <- node개수
# # ```````````````
# left palm:
# size: 61
# #: 1
# tongue:
# size: 33 1
# #: 1 28
# ______________________________
# Global network properties <- 네트워크의 연결성, 견고함 등을 나타냄
# `````````````````````````
# Largest connected component (LCC):
# left palm tongue
# Relative LCC size 1.00000 0.54098
# Clustering coefficient 0.65197 0.57913
# Modularity 0.12369 0.12694
# Positive edge percentage 49.54338 59.89583
# Edge density 0.47869 0.36364
# Natural connectivity 0.12729 0.10811
# Vertex connectivity 10.00000 2.00000
# Edge connectivity 10.00000 2.00000
# Average dissimilarity* 0.84269 0.86923
# Average path length** 1.04898 1.13962
#
# Whole network:
# left palm tongue
# Number of components 1.00000 29.00000
# Clustering coefficient 0.65197 0.57913
# Modularity 0.12369 0.12694
# Positive edge percentage 49.54338 59.89583
# Edge density 0.47869 0.10492
# Natural connectivity 0.12729 0.04686
# -----
# *: Dissimilarity = 1 - edge weight
# **: Path length = Units with average dissimilarity
#
# ______________________________
# Clusters <- 각 네트워크의 cluster 수와 그 안의 노드 개수
# - In the whole network
# - Algorithm: cluster_fast_greedy
# ````````````````````````````````
# left palm:
# name: 1 2 3
# #: 19 23 19
#
# tongue:
# name: 0 1 2 3
# #: 28 10 13 10
#
# ______________________________
# Hubs <- 허브 노드(아래 네가지 방법에서 공통적인 taxa)
# - In alphabetical/numerical order
# - Based on empirical quantiles of centralities
# ```````````````````````````````````````````````
# left palm tongue
# Fusobacterium Campylobacter
# Leptotrichia Leptotrichia
# Oribacterium Unclassified Lachnospiraceae
#
# ______________________________
# Centrality measures
# - In decreasing order
# - Centrality of disconnected components is zero
# ````````````````````````````````````````````````
# Degree (unnormalized):
# left palm tongue
# Unclassified Streptophyta 47 10
# Leptotrichia 47 20
# Bacteroides 46 10
# Unclassified Bacteria 46 0
# Fusobacterium 45 15
# ______ ______
# Leptotrichia 47 20
# Campylobacter 36 20
# Oribacterium 39 20
# Haemophilus 24 18
# [Prevotella] 10 18
#
# Betweenness centrality (normalized):
# left palm tongue
# Fusobacterium 0.05085 0.02419
# Leptotrichia 0.04181 0.0504
# Bacteroides 0.02881 0.01008
# Unclassified Streptophyta 0.02825 0.00403
# Parvimonas 0.02825 0
# ______ ______
# Oribacterium 0.02316 0.14516
# Haemophilus 0.00226 0.08468
# [Prevotella] 0.00282 0.05645
# Rothia 0.00734 0.05444
# Leptotrichia 0.04181 0.0504
#
# Closeness centrality (normalized):
# left palm tongue
# Fusobacterium 1.34753 1.20688
# Neisseria 1.3215 1.14746
# Haemophilus 1.31325 1.21906
# Unclassified Bacteria 1.28898 0
# Leptotrichia 1.2852 1.33367
# ______ ______
# [Prevotella] 0.82772 1.34515
# Leptotrichia 1.2852 1.33367
# Unclassified Lachnospiraceae 1.10444 1.28345
# Campylobacter 1.15364 1.27294
# Veillonella 1.11835 1.24568
#
# Eigenvector centrality (normalized):
# left palm tongue
# Fusobacterium 1 0.84707
# Leptotrichia 0.99148 1
# Oribacterium 0.8844 0.79039
# Unclassified Bacteria 0.86111 0
# Unclassified Streptophyta 0.8609 0.21985
# ______ ______
# Leptotrichia 0.99148 1
# Unclassified Lachnospiraceae 0.61801 0.91662
# Campylobacter 0.80563 0.89528
# Fusobacterium 1 0.84707
# [Prevotella] 0.13195 0.83545
- 왼손바닥의 node수는node 수는 61, 혀에서 single tone을 제외한 node 수는 33개로 구성된다
- 왼손바닥은 3개의 cluster를 가지며, 혀는 4개의 cluster로 구성된다.
- 왼손바닥은 혀보다 connectivity가 높아 더 견고한 네트워크임을 알 수 있다. 견고한 네트워크는 외부의 영향에도 안정적인 네트워크를 더 잘 구성한다고 해석할 수 있음
- 왼손바닥의 허브노드는 Fusobacterium,Leptotrichia, Oribacterium Unclassified이며, 혀의 허브노드는 Campylobacter, Leptotrichia, Lachnospiraceae이다.
6-3. 레이아웃 설정
# Compute layout
ps_graph <- igraph::graph_from_adjacency_matrix(ps_genus.sparcc$adjaMat1,
weighted = TRUE)
set.seed(42)
ps_lay_fr <- igraph::layout_with_fr(ps_graph)
# Row names of the layout matrix must match the node names
rownames(ps_lay_fr) <- rownames(ps_genus.sparcc$adjaMat1)
6-4. network plot 그리기
# Get phyla names
taxtab <- as(tax_table(ps_genus_renamed), "matrix")
phyla <- as.factor(taxtab[, "Phylum"])
names(phyla) <- taxtab[, "Genus"]
table(phyla)
# Define phylum colors
library(RColorBrewer)
phylcol <- c(brewer.pal(12, "Paired"), brewer.pal(12, "Set3"))
# Colors used in the legend should be equally transparent as in the plot
phylcol_transp <- colToTransp(phylcol, 60)
plot(ps_props_sparcc,
layout = "spring",
nodeSize = "eigenvector", # "clr",
nodeColor = "cluster", #"feature",
rmSingles = T,
cexNodes = 0.8,
cexHubs = 1.1,
featVecCol = phyla,
colorVec = phylcol,
posCol = "darkturquoise",
negCol = "orange",
title1 = "Network on genus level with sparcc correlations",
showTitle = TRUE,
groupNames = c("left palm","tongue"))
legend(-1.2, 10, cex = 1.5, pt.cex = 2.5, title = "Phylum:",
legend=levels(phyla), col = phylcol_transp, bty = "n", pch = 16)
legend(0.7, 1.0, cex = 2.2, title = "estimated correlation:",
legend = c("+","-"), lty = 1, lwd = 3, col = c("darkturquoise","orange"),
bty = "n", horiz = TRUE)
dev.off()
- 위 그림도 각각의 node색을 phylum에 따라 지정하였으며, node의 크기는 eigenvector에 따라 달라진다.
- 이전 네트워크 그림과 다르게 igraph로 만든 레이아웃이 아닌 "spring"을 사용하였다.
네트워크 분석 결과, 혀의 마이크로바이옴에 비해 왼손바닥의 마이크로바이옴이 더 다양하고 더 많은 cluster로 구성되며, Genus level에서 각 node에 대한 연결이 견고하다. 왼손바닥의 중심이 되는 genus는 Fusobacterium, Leptotrichia, Oribacterium Unclassified이며, 혀는 Campylobacter, Leptotrichia, Lachnospiraceae임을 알 수 있다.
기타
- 올해 나온 ggCluster는 NetCoMi보다 구동 방법이 쉬워 보이지만, 아직 사용해보지는 않았다. 더 쉬운 패키지를 찾는 다면 추천한다.
- tutorial (중국어)
https://blog.csdn.net/woodcorpse/article/details/125863402
https://blog.csdn.net/weixin_60157921/article/details/129548610
https://zhuanlan.zhihu.com/p/617569543
Reference
- Matloff, N, A User’s Guide to Network Analysis in R. Journal of Statistical Software, Book Reviews, 72(3), 2016, 1–2. https://doi.org/10.18637/jss.v072.b03
- Layeghifard et al., Disentangling Interactions in the Microbiome: A Network Perspectiv. Trends in Microbiology, 2017
- The Genetic Diversity Centre (GDC), Microbiota Data Analysis 2020 Workshop- Microbial Networks, 2020 (https://www.gdc-docs.ethz.ch/MDA/handouts/MDA20_Network_Analysis.pdf)
- https://microbiome.github.io/OMA/network-learning.html#network-analysis-with-netcomi
- Stefanie Peschel, Christian L Müller, Erika von Mutius, Anne-Laure Boulesteix, Martin Depner, NetCoMi: network construction and comparison for microbiome data in R, Briefings in Bioinformatics, Volume 22, Issue 4, July 2021, bbaa290, https://doi.org/10.1093/bib/bbaa290