
- 작성 일시 : 2023-03-06 ~ 2023-03-21
🟦 1. 마이크로바이옴 데이터에서 기능 예측의 필요성

| Shotgun Metegenome vs. Amplicon
- 금액적 차이 : 미생물 유전체의 전체를 조사하는 Shotgun metagenome 시퀀싱은 약 30$, 16S rRNA(Amplicon)의 시퀀싱 가격은 약 5$ 이하이다.
- 분석 차이 : Shotgun 은 많은 컴퓨팅 파워를 필요로 하고, 샘플의 모든 유전체를 읽어옴으로 사람의 미토콘드리아 데이터가 많이 읽히는 단점이 있다. Amplicon은 일부 마커진을 이용한 계통 정보는 알 수 있지만, 해당 미생물의 전체적인 기능에 대한 정보는 얻을 수 없다.
| 기능 예측 프로그램의 필요성
- 가격적인 면에서 Shotgun를 수행하지 않고도 기능을 추정할 수 있디(PICRUSt는 사람 장 마이크로바이옴에서, amplicon데이터로 WGS와 80~85%의 정확도로 기능적인 예측을 가능하게 해 준다.)
- Shotgun Metegenome 데이터는 샘플에 오염에 취약함으로, 적은 바이오매스를 내는 샘플의 경우 결과가 왜곡될 수 있다(피부 조직 마이크로 바이옴의 경우 피부에 서식하는 미생물이 아니라 미토콘드리아가 많이 시퀀싱 된다). 이를 막기 위해 오염이 덜한 amplicon 방식으로 조사 후 기능을 예측하는 방법이 더 나을 수도 있다.
🟦 2. 기능 예측 도구들
지금까지 여러 도구들이 개발되어 왔으며 크게 기능 추론 프로그램과 환경 미생물의 특성 추론으로 나눌 수 있다. 그러나 이 글에서는 기능 추론 도구 중 Tax4Fun과 PICRUSt에만 집중하여 서술하였다.

| 16S rRNA 에서 Metagenome을 추론하는 알고리즘의 등장
- K-Nearest Neighbor approach(2012) : 16S rRNA에서 k-Nearest Neighbor접근법을 이용해 Metagenome을 예측하는 알고리즘을 처음 게재하였다.
| 미생물 기능 예측 프로그램의 시초
- PICRUSt(2013) : OTU기반으로 feature를 구성한 수, Greengene기반으로 만들어진 계통수에 16S rRNA를 위치시킨 후 각 유전체의 copy number와 기능 유전체를 예측한다.
| PICRUSt를 보완하면서 탄생한 기능 예측 도구 들
- CopyRighter(2014) : 이는 16S rRNA에 대한 copy number를 추론하는 도구이다.
- PanFP(2015) : PICRUSt와 비슷하지만 genome 예측은 각 계통 그룹을 참고한다. 장점은 reference에 있는 OTU 뿐만 아니라 모든 OTU를 사용 가능하며, 단점은 진화적 모델이 만들어지지 않고, 신뢰구간이 구성되지 않는다.
- Tax4Fun(2015) : Nearest Neighbor접근법을 사용했으며, SILVA 데이터베이스에 등록된 16S rRNA의 KEGG를 이용하여 기능을 예측하는 도구이다. OTU단위로 예측하여 사용한다.
- PAPRICA(2015) : 이 프로그램도 이미 제작된 계통수에 16S rRNA를 위치시킨 데이터를 이용한다.
- Piphillin(2016) : Second Genome사가 제작한 분석도구로, 16S와 Reference tree와 비교하여 얻어진 nearest-neighbour clustering에 근거하여 메타게놈을 예측한다. 이는 온라인에서 손쉽게 분석 가능하다.
| 기존의 소프트웨어를 보완한 두 번째 버전의 도구들
- Tax4Fun2(2020) : 기존에는 SILVA로 이루어진 데이터베이스에 기초하였으나 다른 데이터도 분석 가능하며, Feature를 구성할 때 OTU 뿐만 아니라 ASV도 사용된다.
- PICRUSt2(2020) : Tax4Fun2와 같이 OTU뿐만 아니라 ASV로 fearture구성이 가능하고, 기존에 Greengene에 제한되었던 계통수가 아니라 자체적인 2만여 개의 서열이 담긴 참고 계통수를 구성하였다.
이 중에서 사람 마이크로바이옴 연구에서 가장 많이 사용되는 도구는 PICRUSt와 Tax4Fun이다.
각 프로그램은 개발된 순서를 따라 PICRUSt - Tax4Fun -> Tax4Fun2 -> PICRUSt2 순으로 작성하였다.

🟦 3. PICRUSt 의 알고리즘
PICRUSt(Phylogenetic Investigation of Communities by Reconstruction of Unobserved States)는 16S rRNA에서 Metagenome 데이터의 기능을 예측하는 생물정보학 소프트웨어이다. 파이썬으로 작성되었으며 리눅스 환경과 온라인에서도 실행이 가능하다.
PICRUSt의 분석 단계는 크게 1) Genome prediction algorithm과, 2) Metagenome prediction algorithm으로 나뉜다.
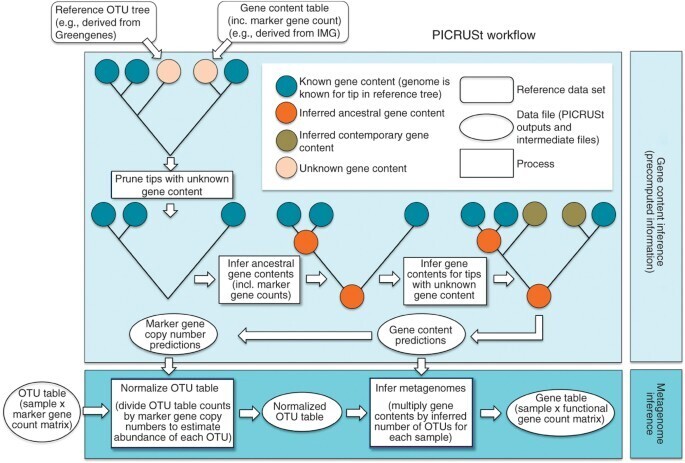
| PICRUSt 분석 단계 1 : Genome prediction algorithm

- input dataset의 quality를 확인한 후, Greengene database의 서열을 기반으로 만들어진 Reference tree에 gene content를 위치시켜 각 gene content의 조상을 파악한다.
- 이때 Reference genome에 매치되지 않은 Unknown gene content는 ancestral state reconstruction (ASR; 총 4가지) 방법을 통해 추론된다.
- 계통적으로 더 가까운 분류군은 가중치가 적용된다.
| PICRUSt 분석 단계 2 : Metagenome prediction algorithm

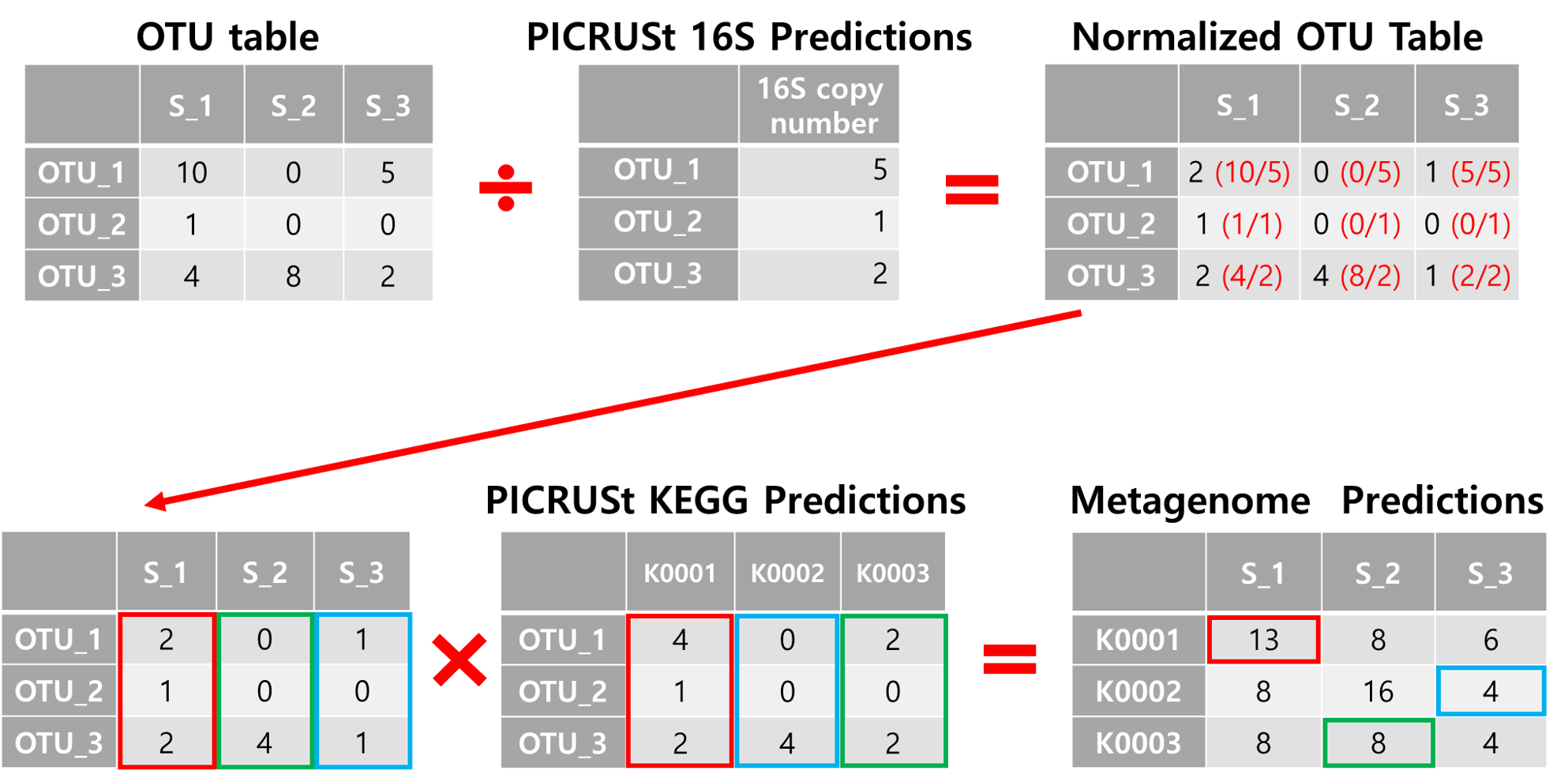
- 파악된 조상 gene contene에서 copy number 수를 예측한 후, OTU table을 각 유전체 수의 copy number로 나누어 normalization한다.
- 이후 Reference tree를 기반으로 예측된 KEGG 정보의 풍부도를 otu 테이블을 곱하여 Metagenime Prediction table을 완성한다.
각 단계를 자세한 데이터로 표현하면 아래와 같다.
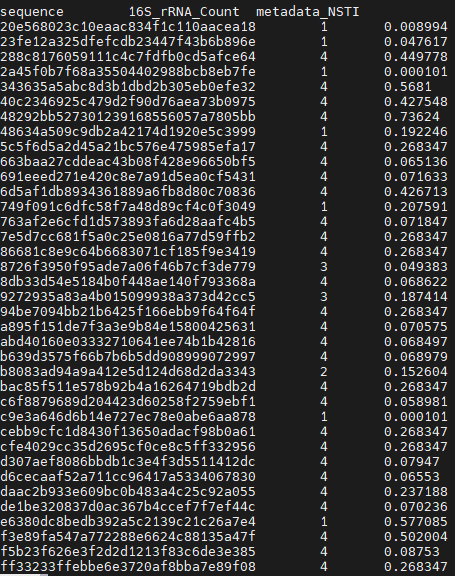
| NSTI score
PICRUSt나 유전적 진화 비교를 할 때 서열을 기반으로 하기 때문에, 잘 연구된 환경의 생물학적 샘플은 다른 환경의 샘플보다 예측이 잘 된다. 예를 들어 과염수 환경보다 사람의 장 미생물 데이터를 사용하였을 때 PICRUSt의 정확도가 높다. 이러한 정확도를 측정하기 위해 PICRUSt는 NSTI값을 사용하였다.
NSTI는 Nearest Sequenced Taxon Index의 약자로, 샘플의 16S rRNA 데이터와 GreenGenes기반의 Reference tree에서 가장 가까운(Nearest) 서열과의 유사성을 측정하는 지표이다. 샘플의 16S rRNA 데이터와 GreenGenes에 있는 미생물의 차이를 NSTI 값으로 계산한다. 이 값은 Reference tree에서 샘플의 16s rRNA값과 유사한 서열 간의 branch를 평균 낸 값이다.
NSTI값은 0부터 1까지의 범위에서 측정되며, 0에 가까울수록 예측된 미생물 군집과 가장 유사한 서열이 데이터베이스에 등록된 미생물 군집과 일치하는 것을 뜻한다. 예를 들어, 샘플 A의 NSTI 값이 0.05라면, 이는 샘플 A의 16S rRNA 데이터와 Greengenes기반의 Reference tree에 등록된 서열 중 가장 유사한 군집과의 차이가 5%라는 것을 의미한다. NSTI 값이 0.2 이상일 경우 noise일 가능성이 높다고 저자는 전했다.
🟦 4. Tax4fun의 알고리즘
- Tax4fun은 R언어로 작성된 패키지이며, R환경과 리눅스 환경에서도 구동 가능하다.
- PICRUSt에 사용된 Greengene은 2013년도 이후로 업데이트가 없으며, KEGG (ver 71.1) reference를 사용하기엔 오직 2,982개의 완전한 원핵 유전자만 가지고 있다. Tax4fun는 PICRUSt보다 많은 양의 (3,808,884 rRNA )유전자 정보를 가진 SILVA SSU databse를 선택하였다.
- Tax4Fun의 논문 저자는 PICRUSt는 nearest neighbor를 기반으로 하기 때문에 실제로 OTU 간의 계통적 길이가 멀어도 모든 OTU가 연결되어 있는 계통수를 가지게 되고, 이는 문 수준에서 거리가 먼 데이터가 많을 때 문제가 될 수 있다고 전했다.
각 분석단계를 PICRUSt와 비교해서 알아보자.
| Tax4Fun 분석 단계 1 : SILVA-based 16S profile is transformed to a taxonomic profile of the KEGG organisms
- PICRUSt의 경우 먼저 Greengene databases를 기반으로 하는 Reference tree에 샘플의 16S rRNA서열로 만들어진 OTU를 위치시켜 Metegenome 서열을 추론한다
- Tax4Fun은 16S rRNA서열로 OTU를 구성하는 단계는 같지만, 이를 Reference tree와 비교하는 것이 아니라, SILVA의 데이터베이스와 비교하여 어떤 학명인지 추론한다(assign). 이 라벨링 된 데이터를 KEGG에 저장된 원핵생물의 whole genome 유전체 정보로 변환한다. 이 변환은 미리 계산된 연관 행렬(SILVA와 KEGG의 정보가 각각 연결된 행렬)에 의해 실행된다.
Table. 연관행렬 (Association Matrix)
| K00001 | K00002 | K00003 | |
| OTU_00001 | 0.0 | 0.001 | 0.0 |
| OTU_00001 | 0.0 | 0.0 | 0.0 |
| OTU_00003 | 0.001 | 0.0 | 0.001 |
행렬은 아래와 같은 단계로 만들어졌다.
1. KEGG(64.0)의 데이터를 BLASTN을 이용하여 계통수를 알아내고 이를 다시 SILVA SSU Ref NR database(115)에서 찾아본다. 각 일치된 정보로 행렬을 만든다.
2. 만들어진 희소행렬은 0이 아닌 항목에 대하여 SILVA에 서열에 KEGG 유기체 중 하나를 할당한다. 이때 k개의 서로 다른 KEGG유기체가 동시에 SILVA서열에 할당될 수 있다. 이때 각 행렬의 값은 1/K값으로 적용된다.
| Tax4Fun 분석 단계 2 : Normalized by the 16S rRNA copy number
- PICRUSt는 Greengene에 저장되어 있는 copy number정보를 사용하는 것이 아니라, 16S rRNA 데이터를 이용하여 copynumber를 예측한다.
- Tax4Fun은 KEGG 유전체 정보를 NCBI genome(RefSeq)과 비교하여 16S copy number 정보를 가져오고, 이 값으로 abundance를 수정한다. 이 방식은 NCBI에 등록된 모든 균주의 copynumber 정보를 사용할 수 있음으로, PICRUSt보다 더 정확한 copynumber 추정을 가능하게 한다.
| Tax4Fun 분석 단계 3 : Combine the precomputed functional profiles of the KEGG organisms for the prediction
- PICRUSt는 Greengenes에 저장된 KEGG Orthology 정보를 이용하여 각 OTU에 맞는 기능 유전체를 예측한다.
- Tax4Fun은 KEGG의 모든 원핵 유전체에 대해 미리 계산된 기능 유전체 데이터와 결합한다. functional reference profiles을 계산하기 위해 UProC(Meinicke, 2014)와 PAUDA(Huson and Xie, 2014)를 사용되며, 박테리아와 고균에서 기원한 총 6977 KO가 저장되어 있다. 이 과정에서 KEGG에 저장된 전장 유전체 서열을 각각 2배의 coverage를 가지는 overaping read로 조각화한다. 이는 기능 유전체로 변환하는 과정에서 생길 수 있는 오류를 최소화하기 위함이며, 긴 길이의 서열의 경우 200bp가 중복되는 400bp길이의 중복된 조각을 생성하고, 짧은 길이의 서열의 경우 50bp가 중복되는 100bp의 조각을 생성한다. 이후 KEGG의 기능 서열들과 대조하여 metagenome의 기능을 예측한다.
| FTU, FSU
FTU(Fraction of OTUs)은 KEGG organisms(KEGG에 등록된 원핵생물 전장 유전체)와 일치하지 않는 OTU를 말한다. 이 값은 어떤 미생물 군집이 많이 발견되었는지 파악할 수 있으며, 이를 통해 샘플의 특성을 파악할 수 있다.
Table. FTU정보를 담고 있는 표
| OTU | 미생물 군 | FTU |
| OTU1 | Bacteroides spp. | 0.25 |
| OTU2 | Prevotella spp. | 0.15 |
| OTU3 | Faecalibacterium spp. | 0.10 |
| OTU4 | Escherichia coli | 0.05 |
| OTU5 | Lactobacillus spp. | 0.02 |
| OTU6 | Streptococcus spp. | 0.01 |
| OTU7 | 기타 미생물 군집 | 0.42 |
FSU( Fraction of sequences unexplained)는 KEGG organisms 서열 중에서 KEGG pathway와 비교하여 일치하지 않은 서열의 비율을 말한다. FSU값이 낮을수록 예측이 정확한 것이다.
Table. FSU정보를 담고 있는 표
| KEGG Orthology ID | Annotation | FSU |
| K00001 | Alcohol dehydrogenase | 0.05 |
| K00002 | Glucose-6-phosphate dehydrogenase | 0.10 |
| K00003 | Dihydroorotate dehydrogenase | 0.02 |
| K00121 | Cytochrome c oxidase subunit I | 0.08 |
| K00123 | Cytochrome c oxidase subunit III | 0.12 |
| K00457 | Glutathione S-transferase | 0.03 |
| KXXXXX | 기타 생물군집 | 0.60 |
두 값을 같이 고려하여서 Tax4Fun의 정확도를 예측할 수 있다. 만약 한 샘플에서 FTU값이 0.5이고 FSU값이 0.1인 경우, 이는 해당 샘플에서 분석된 모든 유전자 서열 중 50%가 KO에 속하며, 예측된 정보의 정확성이 높은 것을 나타낸다.
| WGS vs Tax4Fun vs PICRUSt

WGS데이터와 16S rRNA데이터를 각각의 기능 예측 도구의 결과와 상관관계를 계산한 그래프이다.
- SILVAngs + TAx4Fun은 SILVA의 온라인 데이터 분석 플랫폼에서 계산한 결과이다. 이를 보면 미생물 데이터 분석 프로그램에 상관없이 Tax4Fun을 사용하면 PICRUSt보다 상관관계가 높은 것을 볼 수 있다.
- 추가적으로 PICRUSt와 비교하여 Tax4Fun은 R에서 시각화와 같이 편리하게 사용할 수 있다는 장점이 있다.
- PICRUSt의 NSTI같이 Tax4Fun은 FTU와 FSU같은 신뢰도를 제공한다.
| Tax4Fun vs PICRUSt
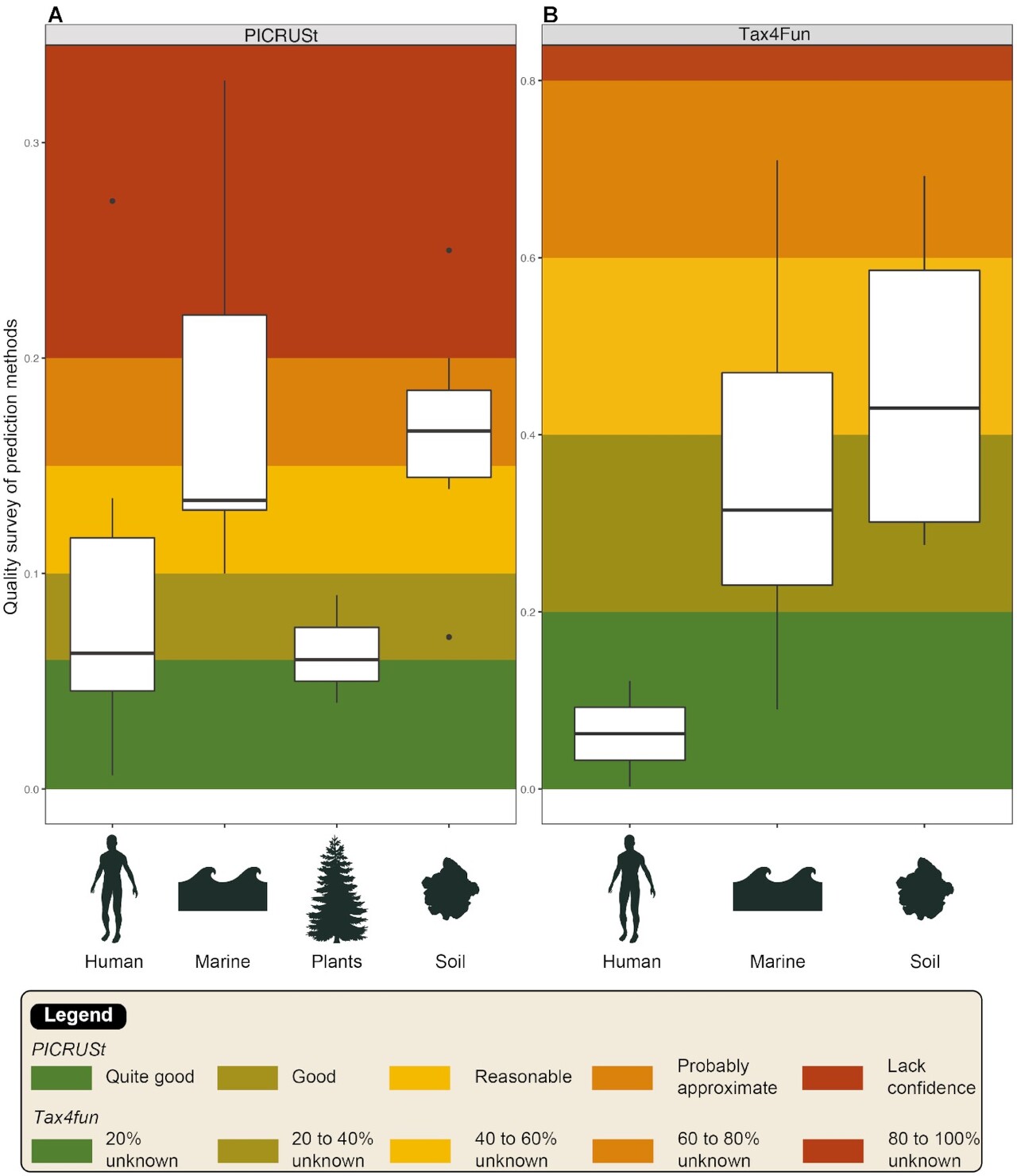
PICRUSt와 Tax4Fun에 대하여 기능 예측의 퀄리티를 측정해 보았다.
PICRUSt는 NSTI기준을 사용(<0.06, quite good; 0.06–0.10, good; 0.10–0.15, reasonable but probably approximate; and >0.20, probably unreliable)하고, Tax4Fun은 KEGG database에 매칭되지 않는지 아닌지를 를 나타내는 FTU를 기준으로 하였다.
둘 다 사람의 데이터는 퀄리티가 높았지만, 환경 데이터로 갈수록 퀄리티가 좋지 않았다.
🟦 5. Tax4fun2의 알고리즘
| Tax4fun2에서 달라진 점
1) Silva뿐만 아니라 사용자가 만든 database도 사용 가능하다.
- 사용자가 원하는 database를 만들고 그 서열의 functional annotation을 평가하기 위해서 extractSSU() 과 assignFunctions() 단계를 추가하였다. 이 단계에서 사용자의 16S rRNA or 18S rRNA는 SILVA SSURef database version 132 를 사용하는 BLAST를 통해 식별된다. Functional profiles은 KEGG 데이터 베이스를 BLASTq를 통해 만들어 낸다. 또한 Protein sequences는 functional annotation전에 prodigal version 2.6.3 을 사용하여 예측된다.
- 현재, functional annotation는 오직 원핵생물만 가능하지만 추가적으로 진핵생물을 위한 버전도 출시할 것이다. 추출된 rRNA서열과 functional profiles은 차후 addUserDataByClustering or addUserData functions 을 이용하여 Reference data set을 만드는데 활용된다.
2) OTU 뿐만 아니라 ASV도 지원한다.
- USEARCH의 UCLSUT알고리즘을 이용하여 99, 100%의 유사성으로 서열을 군집화 한 후, NCBI의 RefSeq database와 비교(mapping, Assignment)한다.
3) SILVA (16S rRNA)를 기반으로 만든 계통수가 사용된다. 만들어진 계통수는 기능적 중복성을 계산하기 위해 사용된다.

| Tax4fun2 분석 단계 1 : 16S rRNA are searched by BLAST (runRefBlast)
- 16S rRNA 유전자는 OTU혹은 ASV 단위로 군집화된 수 BLAST를 통해 Whole metagenome을 알아낸다.
- 이때 유사도 97%를 넘지 않으면 기능예측이 고려되지 않는다.
| Tax4fun2 분석 단계 2 : 기능유전체 예측 (makeFunctionalPrediction)
- OTU table supplied by the user is summarized based on the results of the next neighbour search.
- 기능적 profile을 담고 있는 연관행렬(association matrix)과 통합된다.
| WGS vs Tax4Fun2 vs PICRUSt

| Functional redundancy index
- Tax4Fun2는 PICRUSt와 다르게 기능이 중복될 가능성을 고려하는 계산 단계를 추가로 만들었다. 계산된 값은 FRI(Functional Redundancy Index)로, 다양성과 기능 중복성을 고려하여 기능적 다양성을 나타내는 지수이다. FRI 값은 0에서 1까지의 범위를 가지며, 1에 가까울수록 기능 다양성이 높은 것을 나타낸다.
- FRI = (number of unique reference sequences aligned to the sample dataset) / (total number of reference sequences in the reference dataset)로 예시 데이터와 같이 계산해 보자
만약 reference data가 5000 개의 서열을 가지고 있고, 우리의 샘플 dataset이 총 3000개의 reference와 일치하는 서열을 가지고 있다면 FRI는 3000/5000 = 0.6 값이다. 값이 높을 수록 해당 샘플에서 중복된 기능이 적고, 다양한 기능이 발현되었음을 시사한다.
🟦 6. PICRUSt 2의 알고리즘

| PICRUSt과 PICRUSt2의 차이점
기존 PICRUSt의 단점
1) Closed-reference OTU-picking (기존에 만들어진 16s rRNA의 Database를 기준으로 Align 하여 OTU를 분별)에서 OTU를 생성(즉 de novo가 아니라 reference를 기반으로 하기 때문에 reference게 없는 종은 식별되지 않음 ).
2) 이러한 제한 때문에 PICRUSt의 workflow는 amplicon sequence variants(ASVs)을 만들어내는 sequence의 denoisong 방법과 호환되지 않았다.
3) 또한 PICRUSt가 사용했던 Reference는 Greengene으로 2013년 이후 업데이트 되지 않았다.
PICRUSt2에서 달라진 점
1) PICRUSt2 알고리즘(Fig.1a)은 OTU reference에 의지하기보다는(Fig.1b) 더 많은 양의 데이터(Fig.1c)를 기반으로 하는 reference phylogeny에 서열을 위치시켜 유전자 예측을 최적화하고
2) 좀 더 엄격하게 pathway의 풍부도를 예측(Supplementary Fig. 1)하고 맞춤형 데이터베이스의 통합을 가능하게 한다.
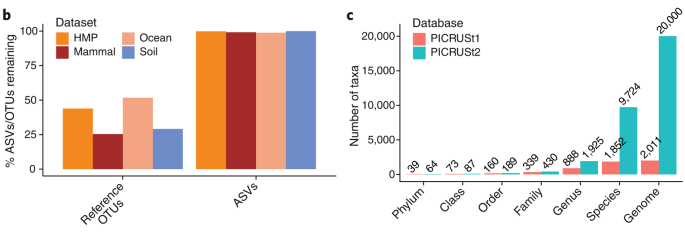
위 논문에도 각 기능 예측 프로그램 간의 비교를 실시하였는 데 사용된 모델은 아래와 같다.
- Null, Taxa4Fun2, PanFP, Piphillin, PICRUSt1, PICRUSt2

a. 각 기능 유전체의 예측 프로그램 간의 Spearman 상관계수를 비교한 결과
- 데이터는 총 7가지이며, 이 중 Indian데이터에서 Piphilion과 PICRUSt2를 제외하고는 다들 유의성 있는 차이를 보였다.
b. 각 Whole genome데이터와 16S amplicon 데이터를 이용해 기능예측을 한 결과를 여러 지수로 측정한 결과이다.
- 여기서 "Shuffled ASVs"는 rASV 레이블을 섞어 높은 데이터이며, "Alt.MGS"는 HUMAn2를 사용하지 않고 KEGG데이터 베이스에서 align 된 데이터이다.
- F1 score와 Recall 지표에서는 PICRUSt2가 대부분 높은 정확도를 보였으며, Precision에서는 Alt.MGS와 Piphillin의 정확도가 높게 측정되었다. 그러나 Piphillin은 Indian데이터의 Recall 평가모델에서 정확도가 매우 낮게 측정되었다.
🟦 7. 비교 : PICRUSt vs. Tax4Fun
by Djemiel C et al
| Tax4FUN | Tax4FUN2 | PICRUSt | PICRUS2 | |
| Feature | OTU | OTU/ASV | OTU | OTU/ASV |
| 작성 언어 | R | R | Python | Python, R |
| marker gene | 16S rRNA | 16S rRNA | 16S rRNA | 16S/18S rRNA/ITS |
| 기능 예측 database | - KO | - KO | - KO - KEGG Pathway - COG - CAZy |
- MetaCyc - KO - EC number - COG - Pfam - TIGRFAM |
| maker gene을 이용한 metagenome 서열 예측 알고리즘 (Copy number 추정) |
Nearest-neighbor search based on a minimum 16S rRNA sequence similarity |
BLAST (NCBI RefSeq) |
ASR (Wagner Parsimony, ACE ML, ACE REML, ACE PIC) | HSP (maximum parsimony, empirical probabilities, subtree averaging, SCP) |
| 사용된 데이터 | Only in SILVA | rDNA amplicon sequences |
Only in Greengenes |
rDNA amplicon sequences |
| 강점 | - 최소 서열 유사도 알고리즘 사용 - R에서 편하게 사용할 수 있다. - 신뢰도가 계산된다 (FTU and FSU) - 특정 기능에 대해 중복을 계산한다 |
- R에서 사용 가능하다 - 신뢰도가 계산된다 (FTU and FSU) - 계통간 거리가 먼 집단에서 기능예측을 더 잘 수행한다 |
- 진화적 모델이 고려된다 - NSTI를 이용한 신뢰도 계산 - OTU의 copynumber를 통한 수정 |
- 진화적 모델이 고려된다 - NSTI를 이용한 신뢰도 계산 - 여러 HSP 방법이 추가된다(branch length의 가중치가 고려된다). |
| 단점 | - SILVA database 에 의존한다 |
- 진핵생물에서는 사용할 수 없다 |
- Greengenes database 에 의존한다 - 진핵 유전체는 적용하기 어렵다 |
- 일부 clade에 대해 rRNA amplicon이 reference tree에 잘못 위치되어 에러가 나타날 수도 있다. |
🟦 미생물 기능 예측 프로그램의 한계
Djemiel(2022)에 따르면 수평 유전자 전달(horizontal gene transfer)이 고려되지 않고, 유전자 손실이나 새로운 유전자의 탄생이나 중복 또한 고려되지 않는다. 수평 유전자 전달률은 계통에 따라 매우 차이 나기 때문에 이러한 고려를 반영하기는 매우 어렵다고 했다. 미생물은 플라스미드 전달을 통해서 여러 기능을 얻을 수 있으며 빠른 적응을 가능하게 하는 DNA 분자이다. 그러나 이 기능 예측에 관한 문헌 정보는 많지 않다. 이는 환경 적응을 위해 계통적으로 멀리 떨어진 개체군 간 이동이 가능하다.
또한 기술적인 관점에서 amplicon seqeuncing의 정확도를 신뢰하기 어렵다. 16S rRNA full length가 가장 좋은 대안이 될 수 있을 것이라고 전했다. 또한 전체 샘플에서 토양, 바다에 비해 사람샘플에서의 퀄리티가 높고, 미생물 중에서는 박테리아가 진핵미생물에 비해 퀄리티가 높다. 이에 대해 저장소의 정기적인 검수와 업데이트가 필요할 것이라고 말했다.
🟦 용어
# MetaCyc
MetaCyc는 생물체의 대사 경로, 생물학적 반응, 효소 및 대사 생성물의 관계에 대한 상세 정보를 제공하는 고급 대사 경로 데이터베이스이다. MetaCyc는 수동으로 작성되며, 전문가들이 직접 정보를 추출하고 검증단계를 거친다. 하지만 MetaCyc는 대상 생물체 범위가 제한적이며, 대사 경로 이외의 정보를 제공하지 않는다.
# KEGG
KEGG (Kyoto Encyclopedia of Genes and Genomes)는 유전체 정보, 생물학적 경로, 생화학적 반응, 질병, 약물 등에 대한 정보를 제공하는 유전체 정보 데이터베이스이다. 다양한 생물체에 대한 정보를 제공합니다. 따라서 KEGG는 대규모 생물학적 정보를 제공하지만, 데이터의 정확성과 상세함에서는 MetaCyc에 비해 떨어질 수 있다.

# Precision= TP/(TP+FP)
모델이 Positive라고 예측한 결과 중에서 실제 Positive인 비율. 즉, 모델이 Positive라고 예측한 것 중에서 얼마나 정확한지를 나타내는 지표이다. Precision은 False Positive를 최소화하는 것을 목표로 한다.
#Recall = Sensitivity = TP/(TP+FN)
실제 Positive 중에서 모델이 Positive라고 예측한 비율. 즉, 실제 Positive 중에서 얼마나 많은 비율을 모델이 예측할 수 있는지를 나타내는 지표이다. Recall은 False Negative를 최소화하는 것을 목표로 한다.
# F1-score = 2 x precision x recall/(precision + recall)
Precision과 Recall의 조화평균(weight harmonic average)으로, 두 지표의 균형을 나타내는 지표입니다. 이는 재현율과 정확도가 반비례할 높은 가능성을 보정하기 위한 지표이며, 데이터가 불균형할 경우 사용한다. F1-score는 높을수록 분류 모델의 성능이 좋다고 판단된다.
# Coverage
같은 염기서열이 여러 번 등장한 정도를 의미한다. Reference서열과 비교하여서 즉 Coverage가 높은 것은 Reference 서열과 일치하는 read의 수가 많다는 것이다.

🟦 참고
- PICRUSt1 wiki : https://en.wikipedia.org/wiki/PICRUSt
- Aßhauer KP, Wemheuer B, Daniel R, Meinicke P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics. 2015, 17. https://doi.org/10.1093/bioinformatics/btv287.
- Wemheuer F, Taylor JA, Daniel R et al. Tax4Fun2: prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environmental Microbiome, 2020, 15. https://doi.org/10.1186/s40793-020-00358-7.
- Langille M, Zaneveld J, Caporaso J et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol, 2013, 31. https://doi.org/10.1038/nbt.2676
- Douglas GM, Maffei VJ, Zaneveld JR et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol, 2020, 38. https://doi.org/10.1038/s41587-020-0548-6
-Djemiel C, Maron P, Terrat S, Dequiedt S, Cottin A, Ranjard L. Inferring microbiota functions from taxonomic genes: a review, GigaScience, 2022, 11. https://doi.org/10.1093/gigascience/giab090.
- 나의 고마운 친구 ChatGPT
글에 오류가 있다면 마구 지적해주세요! 감사합니다.