
| 함수
metadata의 각 column 별로 x값을 달리해서 그림을 그려주는 함수를 만들어보자. 예제 데이터는 phyloseq의 GlobalPatterns를 사용할 것이다.
추가적으로 GlobalPatterns의 metadata에 "Type"이라는 변수를 추가하여 보자. Type은 각 샘플을 사람 것인지, 환경에서 얻어 온 것인지 혹은 Mock 샘플인지 구분하여 준다.
library(ggpubr)
library(phyloseq)
library(ggplot2)
library(dplyr)
data("GlobalPatterns")
GlobalPatterns
# phyloseq-class experiment-level object
# otu_table() OTU Table: [ 19216 taxa and 26 samples ]
# sample_data() Sample Data: [ 26 samples by 8 sample variables ]
# tax_table() Taxonomy Table: [ 19216 taxa by 7 taxonomic ranks ]
# phy_tree() Phylogenetic Tree: [ 19216 tips and 19215 internal nodes ]
meta <- sample_data(GlobalPatterns)
meta[meta$SampleType %in% c("Feces", "Skin", "Tongue"), "Type"] <- "Human"
meta[meta$SampleType %in% c("Soil", "Freshwater", "Freshwater (creek)", "Ocean","Sediment (estuary)"), "Type"] <- "Environment"
meta[meta$SampleType %in% c("Mock"), "Type"] <- "Mock"
meta
sample_data(GlobalPatterns) <- meta
먼저 분석하기 전에 OTU table을 표준화해준다
Global.rel <- transform_sample_counts(GlobalPatterns, function(x) x / sum(x) )
| Alpha diversity
자동화 함수는 아래와 같다.
[input]
ps = phyloseq
colum_list = 보고자 하는 metadata의 column name
meas = measures like "Shannon", "Simpson"
path = 파일이 저장될 공간
[output]
지정한 Path에 Alpha diversity plot을 저장해 준다
alpha_save_bycolum <- function(ps, colum_list, meas, path) {
for (col in column){
n <- sample_data(ps) %>% data.frame() %>% select(col) %>% unique %>% nrow
plot_richness(ps, x = col , measures=meas, color=col) +
geom_boxplot() +
theme_classic() +
theme(text = element_text(size = 8),
strip.background = element_blank(),
axis.text.x.bottom = element_text(angle = 45,hjust = 1.1,vjust = 0.9),
legend.title = element_blank(),
legend.position = 'none',
axis.title = element_blank())+
if(n <= 2 ){ stat_compare_means(method = "wilcox.test", tip.length=0.01, size = 2)
}else {stat_compare_means(method = "kruskal.test", tip.length=0.01, size = 2) }
ggsave( paste0(path, col, ".png") , width = 4, height = 3, dpi = 300)
}
}실제 사용은 아래와 같다
## plot
column <- c("SampleType", "Type")
path <- "../alpha/"
alpha_save_bycolum(Global.rel, column, "shannon", path)
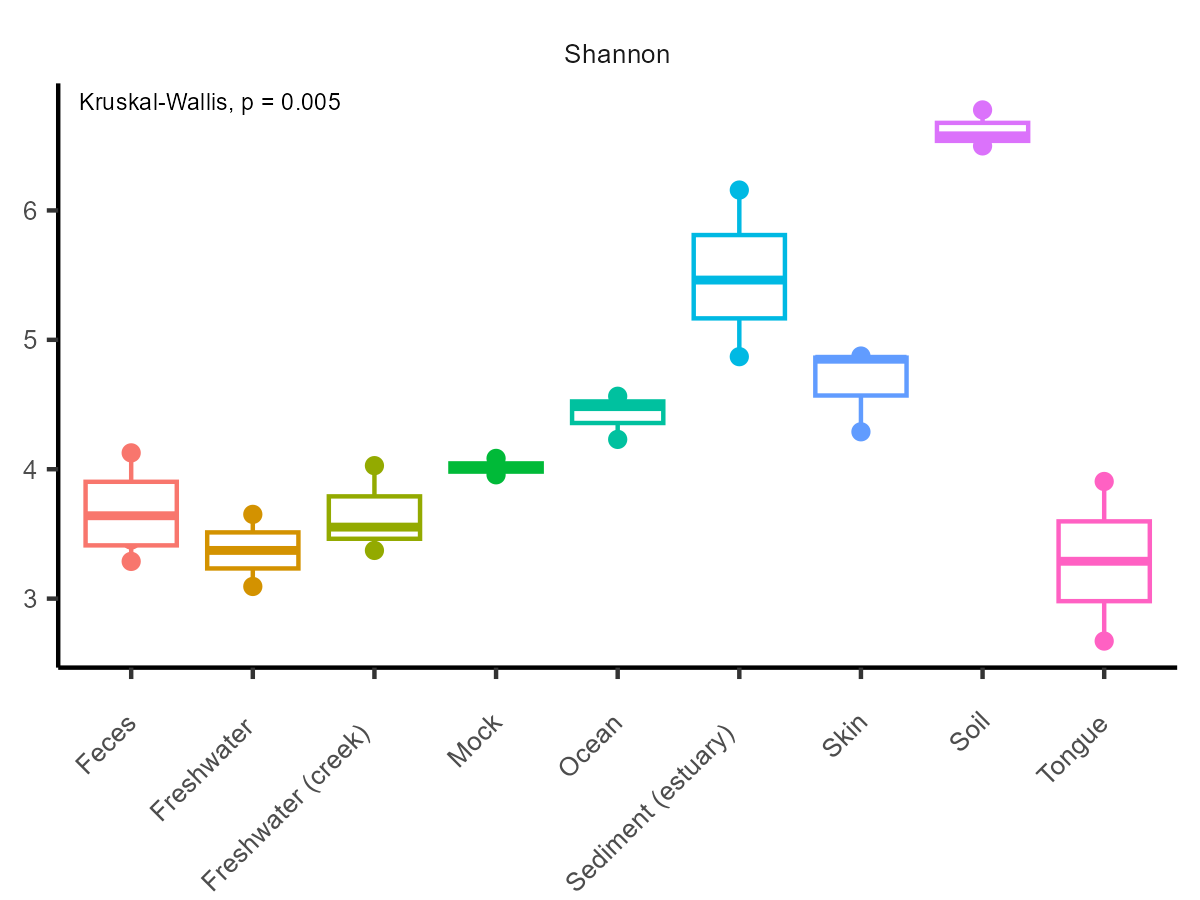

| Beta diversity
[input]
ps = phyloseq
colum_list = 보고자 하는 metadata의 column name
plot = 차원축소 방법 지정 like "PCoA", "NMDS" 등
index = distance matrix 방법 지정 like "bray", "unifrac" 등
path = 파일이 저장될 공간
seed = set.seed() 값
[output]
지정한 Path에 Beta diversity plot을 저장해 준다
library(plyr)
library(reshape2)
library(vegan)
library(glue)
library(ggtext)
Beta_save_bycolum <- function(ps, colum_list, plot, index, path, seed) {
for (col in column){
x.dist <- phyloseq::distance(ps, method = index)
set.seed(seed)
myfunc <- function(v1) { # https://stackoverflow.com/questions/14577412/how-to-convert-variable-object-name-into-string
deparse(substitute(v1))
}
dist <- myfunc(x.dist)
a <- adonis2(as.formula (glue("{dist} ~ {col}")), data=data.frame(sample_data(ps)),permutations=9999, method=index)
pval <- a$`Pr(>F)`[1]
set.seed(seed)
ord <- ordinate(ps, plot, index)
p <- plot_ordination(physeq = ps, ordination = ord, label = NULL, color=col) +
stat_ellipse() + geom_point() +
theme_classic() +
theme(plot.caption = element_text(hjust = 0, size = 8)) +
theme(plot.caption = element_markdown(),) +
if (pval < 0.05) { labs(caption = glue("All comparisons were <span style = 'color: #FF0000'> **significant({pval})**</span> using PERMANOVA at 0.05"))}else { labs(caption = glue("All comparisons were not significant({pval}) using PERMANOVA at 0.05"))
# caption = glue("All comparisons were not significant({pval}) using PERMANOVA at 0.05"))
}
p
ggsave( paste0(path, col, ".png") , width = 5, height = 4, dpi = 300)
}
}실제 사용은 아래와 같다
## plot
column <- c("SampleType","Type")
path <- "../beta/"
Beta_save_bycolum(Global.rel, column, "PCoA", "bray", path, 3103)
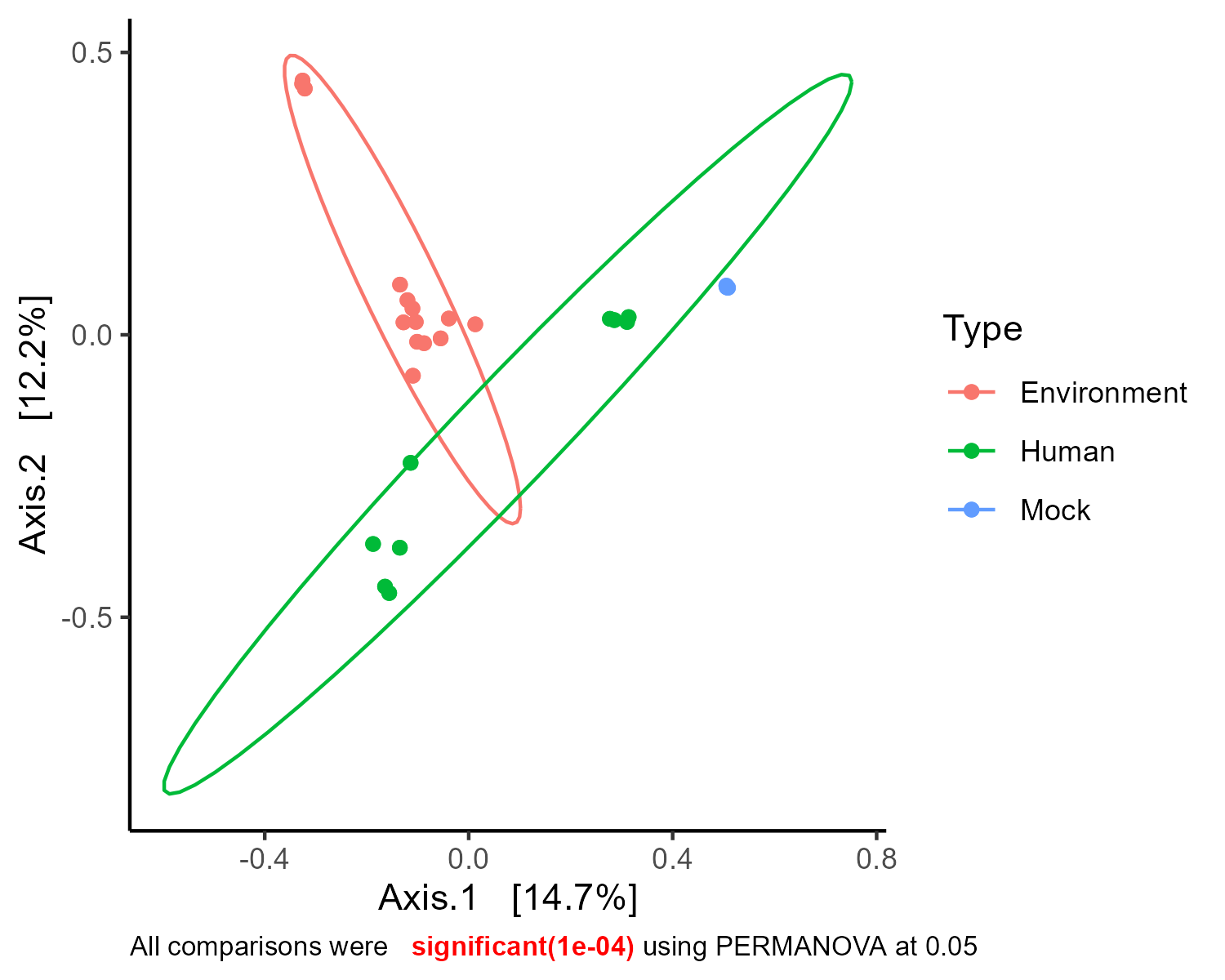
감사합니다.