
작성날짜 : 2023.08.04
수정 : 2023.08.16
- 참고용으로만 작성, 원문을 읽는 것을 추천
- 잘 모르는 부분에 🤔 표시하고 나중에 다시 찾아보기
1. ANCOM-Ⅱ를 써야 하는 이유
최근 논문(Nearing, J.T., 2022)에서 ALDEx2와 ANCOM-Ⅱ가 Microbiome데이터의 differential abundance analysis에 사용하기 적합한 도구라고 소개
2. ANCOM-Ⅱ는 어떻게 marker를 탐지하는가?
- 논문 : https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5682008/
ANCOM-Ⅱ소개
- 마이크로바이옴 데이터의 특징은 0 값이 매우 많다는 것이다. 보통 이때 1 값(pseudo-count)을 더하여 0 값을 없애버리는 보정방법을 많이 사용한다. 그러나 위 논문에서 그 방법은 이상적이지 않다고 저자는 말한다. 또한 probability model을 사용하여 0 값을 처리한다. 그러나 데이터의 유형은 다양함(3가지)으로 모든 데이터에 적용되는 것을 아니다.
ANCOM-Ⅱ의 차별점
1) 0의 유형을 인식하고 3가지로 분류하며
2) 분류된 0의 구조와 유형을 고려하면서 둘 이상의 그룹에서 taxa의 상대적 풍부도를 비교하고
- taxa가 모든 샘플에서 존재한다고 가정. 그러므로 보정값은 모든 샘플에 따라 동일
3) 두 개 이상의 그룹 간에서 제한된 추론을 수행 (여러 실험 조건에서도 폭넓게 사용할 수 있다고 주장)
- cross-sectional/ Repeated measures/ longitudinal designs에서도 사용가능
- 시간 경과 실험, dose-response studies 실험에서도 사용가능
- 여러 실험군과 대조군 간의 multiple pairwise comparisons에서도 사용 가능
참고
n_j : j그룹의 샘플, 논문에서 n으로 적음
j^th : th번째 대조군, 논문에서 j = 1, 2,... J로 적음
i^th : j^th 대조군의 샘플
k^th : i^th 내의 th번째 taxon
z_ijk : k^th의 풍부도
z_ij = j그룹에서 i 샘플의 총 풍부도 z_ij1, …, z_ijp)
y_ij = (y_ij1, …, yij_p) 은 j번째 그룹에서 i번째 샘플에 대한 p 차원 백터를 말한다. 🤔🤔🤔
ANCOM-Ⅱ의 Zero 값 유형
1) Outlier zeros : 대부분에 샘플에서 존재하지만, 특정 샘플에서 존재하지 않는 taxa를 말함
① 먼저, otu data에 pseudo-count(보통 1 값)을 더한 후 정규화(Aittchisons centered log-ratio; ALR)
② 모델링 :
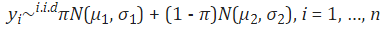
- 이 방법론에서는 두 개의 정규분포가 확연하게 분리되어야 한다.
- 만약 π값이 작은 경우 왼쪽의 cluster가 outlier의 집합이라고 보는 것이 타당하다. 🤔
- 만약 두 그룹이 잘 분리되지 않는다면, 관찰된 zero는 다른 유형의 zero값에 해당할 것이다.
③ 두 정규분포 식별은 아래 기준에 따라 수행
- Frequency : 하나의 분포가 다른 분포에 비해서 c% 더 무거워야 한다. 즉 하나의 정규분포가 전체 데이터의 일부를 차지하는 비율이 c% 이상. c는 π보다 작으며, 미리 정해진 값이다. (위 논문에서는 0.15 값 제)
→ 이때 한 분포가 다른 분포보다 더 많은 비중을 차지하면 outlier zeros로 분류
- Separation : 정규분포 1의 97.5번째 백분위 수와 정규분포 2의 2.55번째 백분위수가 겹치지 않아야 한다.
→ μ₂ - 1.96σ₂ < μ₁ + 1.96σ₁ 일 때, outlier zeros로 분류
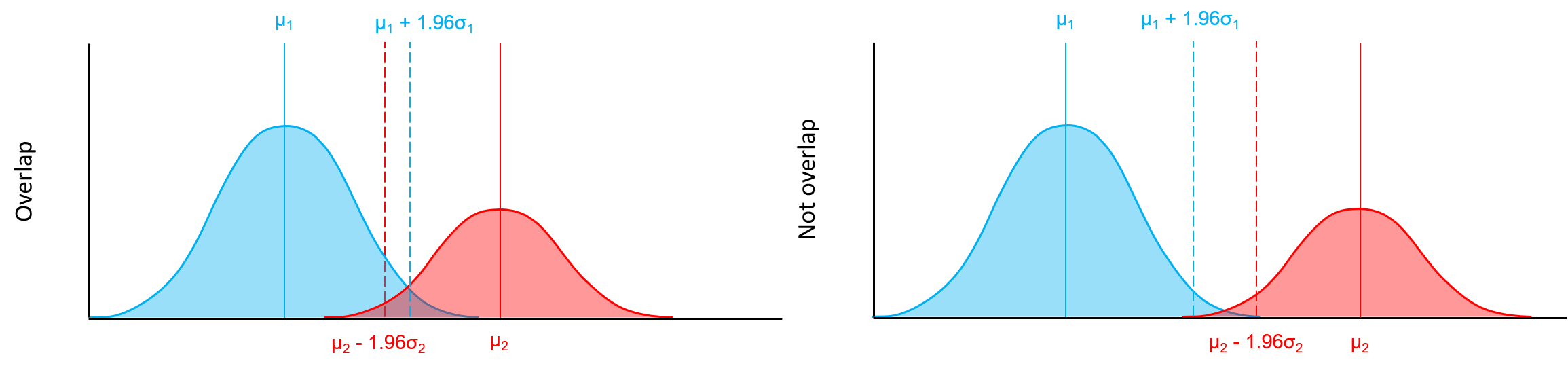
⇒ 결과적으로 outlier zeros로 판별된 값은 NA처리되어 데이터에서 무시된다.
2) Structural zeros : 특정 조건 내에 해당 taxa가 존재하지 않아야 하는 경우를 말함.
- 예를 들어 항생제 노출된 아기의 대변에는, 정상대조군에 존재하는 일부 taxa가 없을 수 있다. 그러나 이론적으로 일부 분류군이 완전히 없을 것으로 예상되지만, 대조군에서 완전히 누락되지 않을 수 있음.
- p = 실험군의 모든 샘플에서 zero값이 아닌 taxa의 비율이라고 가정. 그러나 otu table의 70~80는 모두 zero 값을 가짐. 그렇다면, p는 0 값에 가까울 것이라고 예상.
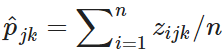
- 그러나 실험군에 존재할 것이라고 예상되지 않는 taxa의 값이 zero가 아닐 때, 이를 structural zeros라고 한다. 즉, 아래 가정이 위반될 때 structural zero라고 한다
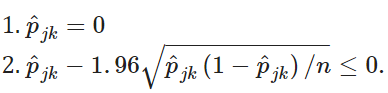
⇒ structural zero값은 이후 data matrix에서 제거
3) Sampling zeros : 표본추출과정에서 우연히 발생하는 Zero값
- ourlier나 strucrue로 구분되지 않는 zero 값을 sampling zeros로 정의한다.
- 다른 분류군에 비해 상대적으로 희귀하거나 기술적으로 관찰되지 않는 taxa에서 관찰된 zero값임
⇒ 이는 pseudo-count(보통 1 값)으로 처리됨
ANCOM-Ⅱ의 가정들 🤔🤔🤔
3. ANCOM-Ⅱ구동 코드
- 코드 출처 : https://github.com/FrederickHuangLin/ANCOM-Code-Archive#repeated-measurelongitudinal-analysis
GitHub - FrederickHuangLin/ANCOM-Code-Archive: Archive: R functions for Analysis of Composition of Microbiomes (ANCOM). Please c
Archive: R functions for Analysis of Composition of Microbiomes (ANCOM). Please check our ANCOMBC R package for the most up-to-date ANCOM function. - GitHub - FrederickHuangLin/ANCOM-Code-Archive: ...
github.com
1. 예제 데이터 불러오기
- QIIME2의 tutorial 데이터
library(phyloseq)
library(readr)
library(tidyverse)
library(ggplot2)
library(ggrepel)
ps <- readRDS("./ps.rds")
# 두 사이트를 비교해기 위해 샘플 추출
ps.gt <- subset_samples(ps, body.site %in% c("gut", "tongue"))
2. 깃허브에서 코드를 다운로드한 후 sourcr("programs/ancom.R")을 통해 코드를 불러온다.
- 아래 코드는 "ancom.R" 코드 일부 수정(figure에서 enrich group에 따라 색으로 달리 하도록 수정)한 코드임
- Standard analysis를 기반으로 작성됨 : 각기 다른 그룹에서 differentially abundant taxa를 감지하고 싶을 때 사용
- 반복 데이터나 공변량을 고려해야 할 때는 깃허브 문서에 맞게 세부 옵션(group_var 등의 변수)을 수정해야 한다.
ANCOM_modi <- function(feature_table, meta_data, tax_data, tax_level = "Species",
struc_zero = NULL, main_var, p_adj_method = "BH",
alpha = 0.05, adj_formula = NULL, rand_formula = NULL, lme_control = NULL){
# OTU table transformation:
# feature_table_pre_process 단계에서 본인의 otu table에 structural zeros가 존재하는지 판별한다
# (1) Discard taxa with structural zeros (if any); (2) Add pseudocount (1) and take logarithm.
if (!is.null(struc_zero)) { # struc_zero가 존재한다면 otu table에 pseudo-count 인 1값을 더하가
num_struc_zero = apply(struc_zero, 1, sum)
comp_table = feature_table[num_struc_zero == 0, ]
}else{ # struc_zero가 없다면 otu table 그대로 사용해라
comp_table = feature_table
}
comp_table = log(as.matrix(comp_table) + 1) # CLR trandf을 위한 log값 변환
n_taxa = dim(comp_table)[1]
taxa_id = rownames(comp_table)
n_samp = dim(comp_table)[2]
# Determine the type of statistical test and its formula. 세부 옵션에 따른 통계적 계산
if (is.null(rand_formula) & is.null(adj_formula)) {
# Basic model
# Whether the main variable of interest has two levels or more?
if (length(unique(meta_data%>%pull(main_var))) == 2) {
# Two levels: Wilcoxon rank-sum test
tfun = stats::wilcox.test
} else{
# More than two levels: Kruskal-Wallis test
tfun = stats::kruskal.test
}
# Formula
tformula = formula(paste("x ~", main_var, sep = " "))
}else if (is.null(rand_formula) & !is.null(adj_formula)) {
# Model: ANOVA
tfun = stats::aov
# Formula
tformula = formula(paste("x ~", main_var, "+", adj_formula, sep = " "))
}else if (!is.null(rand_formula)) {
# Model: Mixed-effects model
tfun = nlme::lme
# Formula
if (is.null(adj_formula)) {
# Random intercept model
tformula = formula(paste("x ~", main_var))
}else {
# Random coefficients/slope model
tformula = formula(paste("x ~", main_var, "+", adj_formula))
}
}
# Calculate the p-value for each pairwise comparison of taxa.
p_data = matrix(NA, nrow = n_taxa, ncol = n_taxa)
colnames(p_data) = taxa_id
rownames(p_data) = taxa_id
pb = txtProgressBar(0, n_taxa - 1, style = 3)
for (i in 1:(n_taxa - 1)) {
setTxtProgressBar(pb, i)
# Loop through each taxon.
# For each taxon i, additive log ratio (alr) transform the OTU table using taxon i as the reference.
# e.g. the first alr matrix will be the log abundance data (comp_table) recursively subtracted
# by the log abundance of 1st taxon (1st column) column-wisely, and remove the first i columns since:
# the first (i - 1) columns were calculated by previous iterations, and
# the i^th column contains all zeros.
alr_data = apply(comp_table, 1, function(x) x - comp_table[i, ])
# apply(...) allows crossing the data in a number of ways and avoid explicit use of loop constructs.
# Here, we basically want to iteratively subtract each column of the comp_table by its i^th column.
alr_data = alr_data[, - (1:i), drop = FALSE]
n_lr = dim(alr_data)[2] # number of log-ratios (lr)
alr_data = cbind(alr_data, meta_data) # merge with the metadata
# P-values
if (is.null(rand_formula) & is.null(adj_formula)) {
p_data[-(1:i), i] = apply(alr_data[, 1:n_lr, drop = FALSE], 2, function(x){
test_data = data.frame(x, alr_data, check.names = FALSE)
suppressWarnings(p <- tfun(tformula, data = test_data)$p.value)
return(p)
}
)
}else if (is.null(rand_formula) & !is.null(adj_formula)) {
p_data[-(1:i), i] = apply(alr_data[, 1:n_lr, drop = FALSE], 2, function(x){
fit = tfun(tformula,
data = data.frame(x, alr_data, check.names = FALSE),
na.action = na.omit)
p = summary(fit)[[1]][main_var, "Pr(>F)"]
return(p)
}
)
}else if (!is.null(rand_formula)) {
p_data[-(1:i), i] = apply(alr_data[, 1:n_lr, drop = FALSE], 2, function(x){
fit = try(tfun(fixed = tformula,
data = data.frame(x, alr_data, check.names = FALSE),
random = formula(rand_formula),
na.action = na.omit,
control = lme_control),
silent = TRUE)
if (inherits(fit, "try-error")) {
p = NA
} else {
p = anova(fit)[main_var, "p-value"]
}
return(p)
}
)
}
}
close(pb)
# Complete the p-value matrix.
# What we got from above iterations is a lower triangle matrix of p-values.
p_data[upper.tri(p_data)] = t(p_data)[upper.tri(p_data)]
diag(p_data) = 1 # let p-values on diagonal equal to 1
p_data[is.na(p_data)] = 1 # let p-values of NA equal to 1
# Multiple comparisons correction. # p-value값 보정
q_data = apply(p_data, 2, function(x) p.adjust(x, method = p_adj_method))
# Calculate the W statistic of ANCOM.
# For each taxon, count the number of q-values < alpha.
W = apply(q_data, 2, function(x) sum(x < alpha))
# Organize outputs
out_comp = data.frame(taxa_id, W, row.names = NULL, check.names = FALSE)
# Declare a taxon to be differentially abundant based on the quantile of W statistic.
# We perform (n_taxa - 1) hypothesis testings on each taxon, so the maximum number of rejections is (n_taxa - 1).
out_comp = out_comp %>% # w 임계값에 따라 통과 여부 표시
mutate(detected_0.9 = ifelse(W > 0.9 * (n_taxa -1), TRUE, FALSE),
detected_0.8 = ifelse(W > 0.8 * (n_taxa -1), TRUE, FALSE),
detected_0.7 = ifelse(W > 0.7 * (n_taxa -1), TRUE, FALSE),
detected_0.6 = ifelse(W > 0.6 * (n_taxa -1), TRUE, FALSE))
# Taxa with structural zeros are automatically declared to be differentially abundant
if (!is.null(struc_zero)){
out = data.frame(taxa_id = rownames(struc_zero), W = Inf, detected_0.9 = TRUE,
detected_0.8 = TRUE, detected_0.7 = TRUE, detected_0.6 = TRUE,
row.names = NULL, check.names = FALSE)
out[match(taxa_id, out$taxa_id), ] = out_comp
}else{
out = out_comp
}
# Draw volcano plot 시각화
# Calculate clr
clr_table = apply(feature_table, 2, clr)
# Calculate clr mean difference
eff_size = apply(clr_table, 1, function(y)
lm(y ~ x, data = data.frame(y = y,
x = meta_data %>% pull(main_var),
check.names = FALSE))$coef[-1])
if (is.matrix(eff_size)){
# Data frame for the figure
dat_fig = data.frame(taxa_id = out$taxa_id, t(eff_size), y = out$W, check.names = FALSE) %>%
mutate(zero_ind = factor(ifelse(is.infinite(y), "Yes", "No"), levels = c("Yes", "No"))) %>%
gather(key = group, value = x, rownames(eff_size))
# Replcace "x" to the name of covariate
dat_fig$group = sapply(dat_fig$group, function(x) gsub("x", paste0(main_var, " = "), x))
# Replace Inf by (n_taxa - 1) for structural zeros
dat_fig$y = replace(dat_fig$y, is.infinite(dat_fig$y), n_taxa - 1)
fig = ggplot(data = dat_fig) + aes(x = x, y = y) +
geom_point(aes(color = zero_ind)) +
facet_wrap(~ group) +
labs(x = "CLR mean difference", y = "W statistic") +
scale_color_discrete(name = "Structural zero", drop = FALSE) +
theme_bw() +
theme(plot.title = element_text(hjust = 0.5), legend.position = "top",
strip.background = element_rect(fill = "white"))
fig
} else{
out_data <- merge(out, tax_data, by.x = "taxa_id", by.y = "row.names")
# Data frame for the figure
dat_fig = data.frame(taxa_id = out$taxa_id, x = eff_size, y = out$W) %>%
mutate(zero_ind = factor(ifelse(is.infinite(y), "Yes", "No"), levels = c("Yes", "No")))
# Replace Inf by (n_taxa - 1) for structural zeros
dat_fig$y = replace(dat_fig$y, is.infinite(dat_fig$y), n_taxa - 1)
# 수정 시작 --
merge_data <- merge(dat_fig, tax_data, by.x = "taxa_id", by.y = "row.names")
counts_tab = clr_table; groups = meta_data[, main_var]; sample_in_cols=T
get_sl_enrich_group <- function(counts_tab, groups, sample_in_cols = TRUE) { # by microbiome Marker
if (sample_in_cols) {
counts_tab <- t(counts_tab)
}
counts_mean <- by(counts_tab, groups, colMeans)
counts_mean <- do.call(cbind, counts_mean)
# idx_enrich <- apply(counts_mean, 1, which.max)
# group_enrich <- colnames(counts_mean)[idx_enrich]
return(data.frame(counts_mean))
}
group_clr_abund <- get_sl_enrich_group(clr_table, meta_data[, main_var], sample_in_cols=T)
merge_data2 <- merge(merge_data, group_clr_abund, by.x = "taxa_id", by.y = "row.names")
gr_compar <- meta_data[, main_var] %>% unique
com1 <- as.character(gr_compar[1])
com2 <- as.character(gr_compar[2])
Minus = merge_data2[, com1] - merge_data2[, com2]
merge_data3 <- merge_data2 %>%
mutate(enrich_group = if_else(Minus >= 0, com1, com2))
# -- 수정 완료
fig = ggplot(data = merge_data3) + aes(x = x, y = y) +
geom_point(aes(color = enrich_group)) + ## 수정
labs(x = "CLR mean difference", y = "W statistic") +
scale_color_discrete(name = "Enrich group", drop = FALSE) +
theme_bw() +
theme(plot.title = element_text(hjust = 0.5), legend.position = "top") +
ggrepel::geom_text_repel(aes_string(label = tax_level), max.overlaps = 15) ## 수정
fig
}
res = list(p_data = p_data, q_data = q_data, out = out_data, fig = fig)
return(res)
}
3. Phyloseq을 사용한 시각화 (Volcano plot) 수행함수
ANCOM_volcano <- function(phyloseq, Group, tax_level, group_var) {
# import
otu_data <- otu_table(phyloseq) %>% as.data.frame() # %>% t() %>% as.data.frame() # absolute abundance table
meta_data <- sample_data(phyloseq) %>% as.data.frame()
tax_data <- tax_table(phyloseq) %>% as.data.frame()
meta_data$SampleID <- rownames(meta_data)
# Step 1: Data preprocessing 데이터 전처리
# 옵션 값 설정
feature_table = otu_data; sample_var = "SampleID"; group_var = NULL
out_cut = 0.05; zero_cut = 0.90; lib_cut = 1000; neg_lb = FALSE
# 전처리
prepro = feature_table_pre_process(feature_table, meta_data, sample_var, group_var,
out_cut, zero_cut, lib_cut, neg_lb)
# 데이터 정리
feature_table = prepro$feature_table # Preprocessed feature table
meta_data = prepro$meta_data # Preprocessed metadata
struc_zero = prepro$structure_zeros # Structural zero info
# Step 2:ANCOM ver 2
# 옵션값 설정
main_var = Group; p_adj_method = "BH"; alpha = 0.05
adj_formula = NULL; rand_formula = NULL; lme_control = NULL;seed = 42
# ANCOM ver 2 분석
set.seed(seed)
res = ANCOM_modi(feature_table, meta_data, tax_data = tax_data, tax_level = tax_level,
struc_zero, main_var, p_adj_method, alpha, adj_formula, rand_formula, lme_control)
res$fig
res$out
# Step 3: Volcano Plot
# Number of taxa except structural zeros
n_taxa = ifelse(is.null(struc_zero), nrow(feature_table), sum(apply(struc_zero, 1, sum) == 0))
# Cutoff values for declaring differentially abundant taxa
cut_off = c(0.9 * (n_taxa - 1), 0.8 * (n_taxa - 1), 0.7 * (n_taxa - 1), 0.6 * (n_taxa - 1))
names(cut_off) = c("detected_0.9", "detected_0.8", "detected_0.7", "detected_0.6")
# Annotation data
dat_ann.7 = data.frame(x = min(res$fig$data$x), y = cut_off["detected_0.7"], label = "W[0.7]")
fig = res$fig +
geom_hline(yintercept = cut_off["detected_0.7"], linetype = "dashed") +
geom_text(data = dat_ann.7, aes(x = x, y = y, label = label), size = 4,
vjust = -0.5, hjust = 0, color = "orange", parse = TRUE)
fig
return(list(out = res$out, fig = fig))
}- otu_table에 따라 t() 삭제 가능
- CLR abundance를 이용해서 각 풍부도의 차이 추정, 그러나 CLR이 높아도 W0.7 이상일 때만 유의한 Marker라고 해석 가능
4. 함수 수행
- Genus level에서
ps.gt <- subset_samples(ps, body.site %in% c("gut", "tongue"))
ps.gt.g <- tax_glom(ps.gt, taxrank = "Genus")
Genus <- ANCOM_volcano(ps.gt.g, "body.site", "Genus")
Genus$fig

W값이 전체에서 70% 이상일 때 유의하다고 판단한다.
- Species level에서
ps.gt.sp <- tax_glom(ps.gt, taxrank = "Species")
Species <- ANCOM_volcano(ps.gt.sp, "body.site", "Species")
Species$fig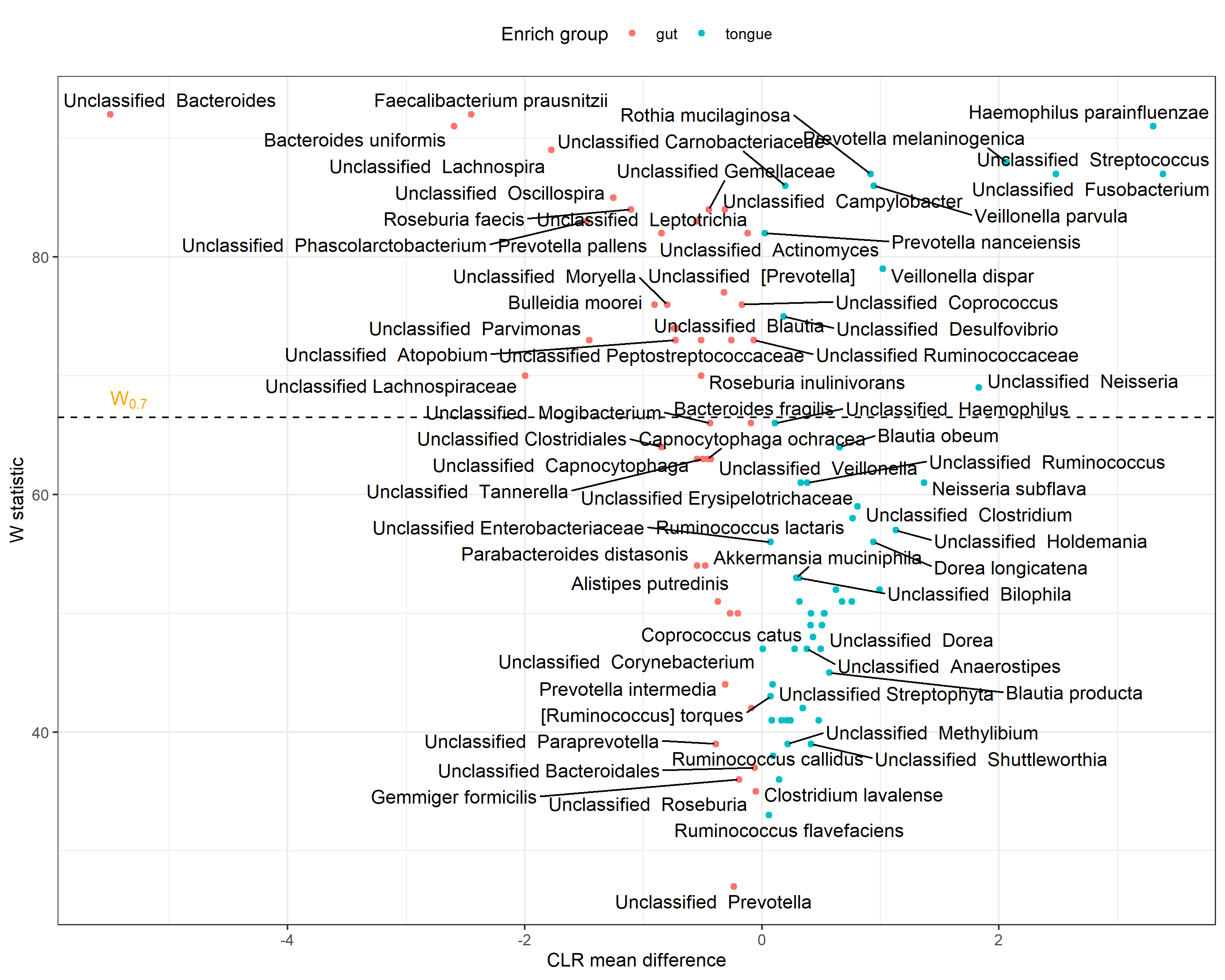
- ASV level에서
ASV <- ANCOM_volcano(ps.gt, "body.site", "Species")
ASV$fig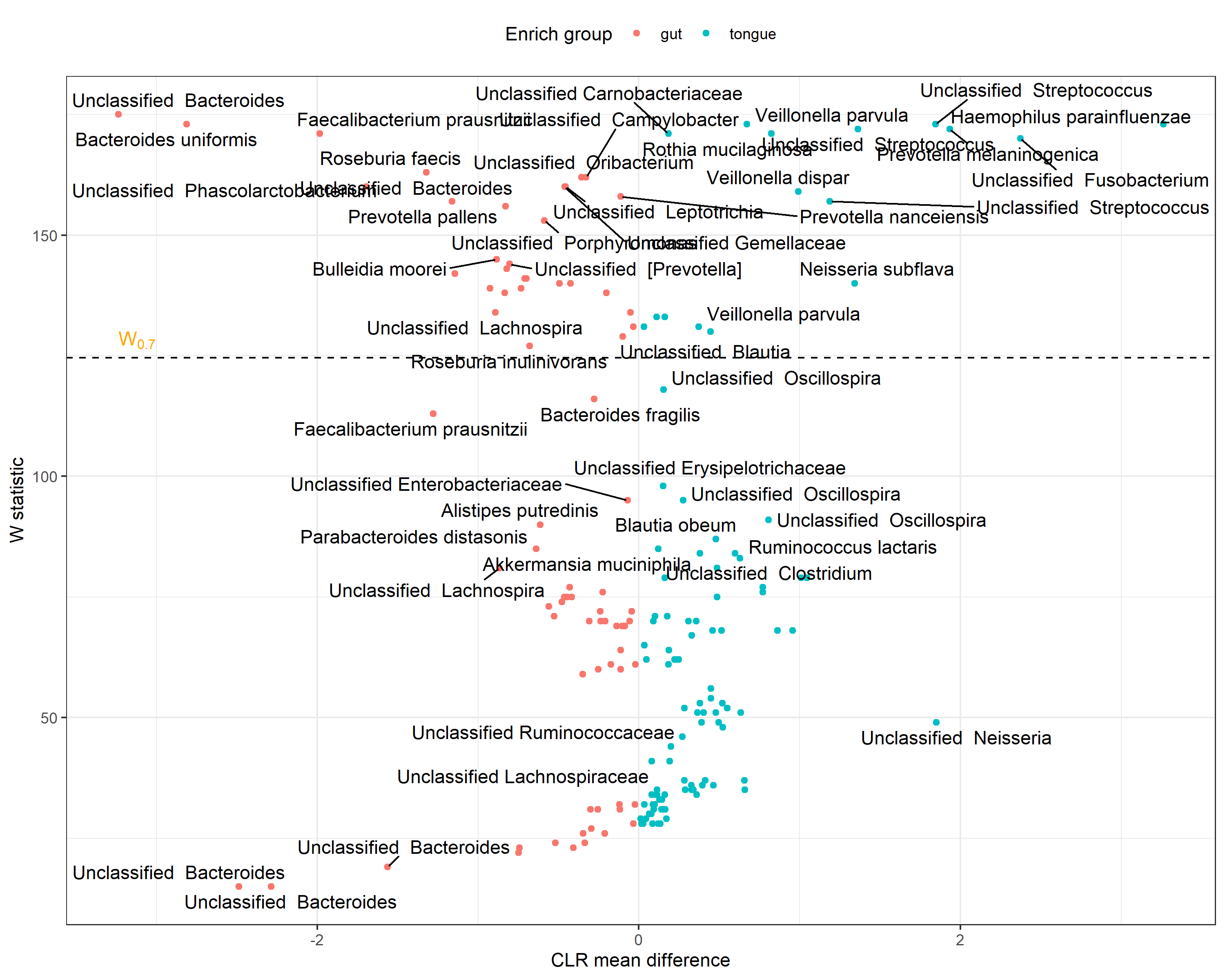
+) ISSUE
실제 데이터에서 lefse, aldex2, ancom2를 수행했을 때 모두 결과 값이 나오지 않았다. ancom2에서 clr difference는 나왔지만 w값이 0인 taxa가 대부분이었다. 이에 대해 몇 가지 문의글을 찾아보았다.
- Issue1 : ANCOM giving me W scores of 0
- Issue2 : all W=0 in ANCOM results
- Issue3 : Another topic of ANCOM - W of 0 - all significant
- Issue4 : ANCOM: 'low W taxa identified as significant' issue's workaround, ANCOM2 code/instructions
패키지 제작자의 답변은 아래와 같다
- 일단 ANCOM ver 2에서 threshold를 수정해서 이러한 현상을 완화함(하지만 나는 2.1 ver에서도 많은 marker가 w=0을 보임)
- 최대한 큰 데이터셋을 사용하라 : Phylum, Genus보다는 Species, ASV level을 사용해라
- Beta diversity에서 통계적으로 유의성을 보였는지 확인해라 : 아니라면, ANCOM에서도 보이지 않을 가능성이 크다