

▶ Dada2란? R을 기반으로 qiime2(리눅스 기반)와 같이 미생물 분석에 사용되는 R 패키지이다.
▶ 이전 글 : [R] Dada2 튜토리얼 ver 1.8 (1)
[R] Dada2 튜토리얼 ver 1.8 (1)
▶ Dada2란? R을 기반으로 qiime2(리눅스 기반)와 같이 미생물 분석에 사용되는 R 패키지이다. ▶ 이전 글 : [R] Dada2 설치 ▶ 튜토리얼 목표 : Dada2로 미생물 데이터 전 처리와 phyloseq을 통한 시각화 ▶
bio-kcs.tistory.com
▶ 튜토리얼 목표 : Dada2로 미생물 데이터 전 처리와 phyloseq을 통한 시각화
▶ 본인의 Dada2 버전에 맞는 튜토리얼을 선택
- Dada2 ver 1.2 tutorial : https://benjjneb.github.io/dada2/tutorial_1_2.html
- Dada2 ver 1.8 tutorial : https://benjjneb.github.io/dada2/tutorial_1_8.html
▶ 참고할 강의 영상 : https://www.youtube.com/watch?v=wV5_z7rR6yw (브라운 대학 워크샵)
▶ 참고할 링크 : https://bioconductor.org/help/course-materials/2017/BioC2017/Day1/Workshops/Microbiome/MicrobiomeWorkflowII.html#graph-based_analyses
▶ 예제 데이터 source : Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. (2013): Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Applied and Environmental Microbiology. 79(17):5112-20.
- 시퀀싱 기계 : illumina MiSeq platform -> paired reads 생성
- 우리가 주목할것 : 박테리아 16s rRNA의 V4영역
🟦 8. Assign Taxonomy
일단 taxonomy를 붙이기 전에 원하는 Database를 다운로드하여야 한다.
dada2는 RDP, Greengene, SILVA모두 사용 가능하며,
위 예제에서는 SILVA ver128을 사용하였다
▶ 데이터 베이스 다운 로드 : https://zenodo.org/record/801832#.Yv2iaHZBzcs
silva_nr_v128_train_set.fa.gz 파일을 원하는 위치에 놓고 그 경로를 아래 넣어 작성하면 된다
taxa <- assignTaxonomy(seqtab.nochim, "~/tax/silva_nr_v128_train_set.fa.gz", multithread=TRUE)
unname(head(taxa))# Removing sequence rownames
#Kingdom Phylum Class Order Family Genus
#[1,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales" "Bacteroidales_S24-7_group" NA
#[2,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales" "Bacteroidales_S24-7_group" NA
#[3,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales" "Bacteroidales_S24-7_group" NA
#[4,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales" "Bacteroidales_S24-7_group" NA
#[5,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales" "Bacteroidaceae" "Bacteroides"
#[6,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales" "Bacteroidales_S24-7_group" NARDP DB가 가장 최신에 업데이트 되어서 많은 사람들이 쓰는 추세이긴 하지만
여기서는 SILVA database를 사용했다
taxa 파일을 살펴보면, Genus level까지만 나와있다
Species level까지 보고 싶다면 아래와 같이 addSpecies() 함수를 이용한다
###Assign species (when possible)
system.time({taxa.plus <- addSpecies(taxa, "~/tax/ silva_species_assignment_v128.fa.gz", verbose=TRUE)
colnames (taxa.plus) <-c("Kingdom", "Phylum", "Class", "Order", "Family", "Genus", "Species")
unname (taxa.plus) })
🟦 9. Evaluate accuracy
unqs.mock <- seqtab.nochim["Mock",]
unqs.mock <- sort(unqs.mock[unqs.mock>0], decreasing=TRUE) # Drop ASVs absent in the Mock
cat("DADA2 inferred", length(unqs.mock), "sample sequences present in the Mock community.\n")
# DADA2 inferred 20 sample sequences present in the Mock community.
mock.ref <- getSequences(file.path(path, "HMP_MOCK.v35.fasta"))
match.ref <- sum(sapply(names(unqs.mock), function(x) any(grepl(x, mock.ref))))
cat("Of those,", sum(match.ref), "were exact matches to the expected reference sequences.\n")
# Of those, 20 were exact matches to the expected reference sequences.이건 나도 무슨 단계인지 모르겠다
일단 Mock 파일과 대조해서 거기 없는 ASV면 제거한다는 뜻인 것 같다
🟦 10.Align Sequence
이제 우리는 이제 otu table과 taxonomy table두 가지 데이터를 가지고 있다
그러나 계통수 파일은 없다!
bray curtis나 shannon지수등을 계산할 때 필요는 없지만, Unifrac지수를 계산하려고 하면 tree파일이 필요하다
tree를 만들기위해 일단 서열을 정렬(Alignment)해준다
BiocManager::install("DECIPHER")
library(DECIPHER)
library(phangorn)
## Extract sequence from Dada2 output
sequences <- getSequences(seqtab.nochim)
names(sequences) <- sequences
## Run sequence alignment (MSA) unsing DECIPHER
alignment <- AlignSeqs(DNAStringSet(sequences), anchor = NA)
#Determining distance matrix based on shared 8-mers:
# |=============================================================================================================| 100%
#
#Time difference of 0.4 secs
#
#Clustering into groups by similarity:
# |=============================================================================================================| 100%
#
#Time difference of 0.17 secs
#
#Aligning Sequences:
# |=============================================================================================================| 100%
#
#Time difference of 3.29 secs
#
#Iteration 1 of 2:
#
#Determining distance matrix based on alignment:
# |=============================================================================================================| 100%
#
#Time difference of 0.04 secs
#
#Reclustering into groups by similarity:
# |=============================================================================================================| 100%
#
#Time difference of 0.1 secs
#
#Realigning Sequences:
# |=============================================================================================================| 100%
#
#Time difference of 1.82 secs
#
#Iteration 2 of 2:
#
#Determining distance matrix based on alignment:
# |=============================================================================================================| 100%
#
#Time difference of 0.04 secs
#
#Reclustering into groups by similarity:
# |=============================================================================================================| 100%
#
#Time difference of 0.09 secs
#
#Realigning Sequences:
# |=============================================================================================================| 100%
#
#Time difference of 0.3 secs이제 tree를 만들어 준다
이때 사용되는 패키지는 phangorn인데 Fasttree라는 패키지도 자주 쓰인다.
#Change sequence alignment output into a phyDat structure
phang.align <- phyDat(as(alignment, "matrix"), type="DNA")
#Create distance matrix
dm <- dist.ml(phang.align)
#Perform Neighbor joining
treeNJ <- NJ (dm) # Note, tip order = sequence order
#Internal maximum likelihood
fit = pml(treeNJ, data=phang.align)
#model: JC
#
#loglikelihood: -15621.78
#
#unconstrained loglikelihood: -1188.264
#
#Rate matrix:
# a c g t
#a 0 1 1 1
#c 1 0 1 1
#g 1 1 0 1
#t 1 1 1 0
#
#Base frequencies:
# 0.25 0.25 0.25 0.25
## negative edges length changed to 0!
fitGTR <- update(fit, k=4, inv=0.2)
fitGTR <- optim.pml (fitGTR, model="GTR", optInv=TRUE, optGamma=TRUE,
rearrangement = "stochastic", control = pml.control(trace = 0)) # Time
왜 계통수를 만들어야 하는가?
Beta diversity에서 Unifrac은 계통수를 기반으로 하기 때문이다
만약 bray-curtis만 본다면 만들기 않아도 된다
neighbor joining(NJ)알고리즘을 통해 GTR+G+l maximum likelihood tree를 만든다.
여기서 GTR+G+I에서
- GTR은 DNA가 시간에 따라 얼마나 변하는가를 수학적 모델로 나타낸 것이며
- G는 여기에 변화하는 확률이 Gamma distrubution을 따르고
- I는 변하지 않는 DNA의 비율 proportion of invariable sites (I)를 가정한 것이다.
neighbor joining(NJ)을 사용한 계통수를 구축했다.
그러나 NJ방식은 unrooted tree임으로 후에 root를 만들어 주어야 한다
🟦 11. Phyloseq 형식으로 데이터 import 하기
phyloseq형식으로 만들기 위해선 4개의 파일이 필요하다
- sample data
- otu table
- tree
- taxonomy table
우리는 sample data파일을 sample이름에서 추출해 와서 만들어 보자
theme_set(theme_bw())
samples.out <- rownames(seqtab.nochim)
subject <- sapply(strsplit(samples.out, "D"), `[`, 1)
gender <- substr(subject,1,1)
subject <- substr(subject,2,999)
day <- as.integer(sapply(strsplit(samples.out, "D"), `[`, 2))
samdf <- data.frame(Subject=subject, Gender=gender, Day=day)
samdf$When <- "Early"
samdf$When[samdf$Day>100] <- "Late"
rownames(samdf) <- samples.out
samdf
# Subject Gender Day When
#F3D0 3 F 0 Early
#F3D1 3 F 1 Early
#F3D141 3 F 141 Late
#F3D142 3 F 142 Late
#F3D143 3 F 143 Late
#F3D144 3 F 144 Late
#F3D145 3 F 145 Late
#F3D146 3 F 146 Late
#F3D147 3 F 147 Late
#F3D148 3 F 148 Late
#F3D149 3 F 149 Late
#F3D150 3 F 150 Late
#F3D2 3 F 2 Early
#F3D3 3 F 3 Early
#F3D5 3 F 5 Early
#F3D6 3 F 6 Early
#F3D7 3 F 7 Early
#F3D8 3 F 8 Early
#F3D9 3 F 9 Early
#Mock ock M NA Early
이제 모두 만들어졌으니 phyloseq 개체를 만들어 보자
# make phyloseq
ps <- phyloseq(otu_table(seqtab.nochim, taxa_are_rows=FALSE),
sample_data(samdf), tax_table(taxa), phy_tree(fitGTR$tree))
#phyloseq-class experiment-level object
#otu_table() OTU Table: [ 232 taxa and 20 samples ]
#sample_data() Sample Data: [ 20 samples by 4 sample variables ]
#tax_table() Taxonomy Table: [ 232 taxa by 6 taxonomic ranks ]
#phy_tree() Phylogenetic Tree: [ 232 tips and 230 internal nodes이렇게 총 otu, taxonomy, metadata, tree 4개의 요소가 합쳐진 phyloseq 개체를 만들었다.
phyloseq을 만들어 두면 한 번의 명령어로 시각화하기 아주 편하다
🟦 12. Root tree로 변환
마지막으로 tree에 root를 만들어 준다
##Currently, the phylogenetic tree is not rooted
##Though it is not necessary here, you will need to root the tree if you want to calculate any phylogeny based
## diversity metrics (like Unifrac)
set.seed (711)
phy_tree(ps) <- root(phy_tree (ps), sample(taxa_names (ps), 1), resolve.root = TRUE)
is.rooted(phy_tree(ps)) # TRUEset.seed(n)을 하지 않으면, 수행할 때마다 값이 달라진다.
만약에 계통수가 필요한 Unifrac이나 Faith'sPD를 만들지 않는다면 이 단계는 필요하지 않다
계통수 파일이 없으면 Shannon, Simpson (alpha diversity), Bray-Curtis Dissimilarity (beta diversity) 지표만 계산 가능하다
🟦 13. Phyloseq으로 시각화해보기
1) Alpha Diversity Metrics
일단 샘플에 포함되어있는 곰팡이 샘플을 제거하고
Shannon과 Simpson 다양성 지수를 비교해 보자
ps <- prune_samples(sample_names(ps) != "Mock", ps) # Remove mock sample
plot_richness(ps, x="Day", measures=c("Shannon", "Simpson"), color="When") + theme_bw()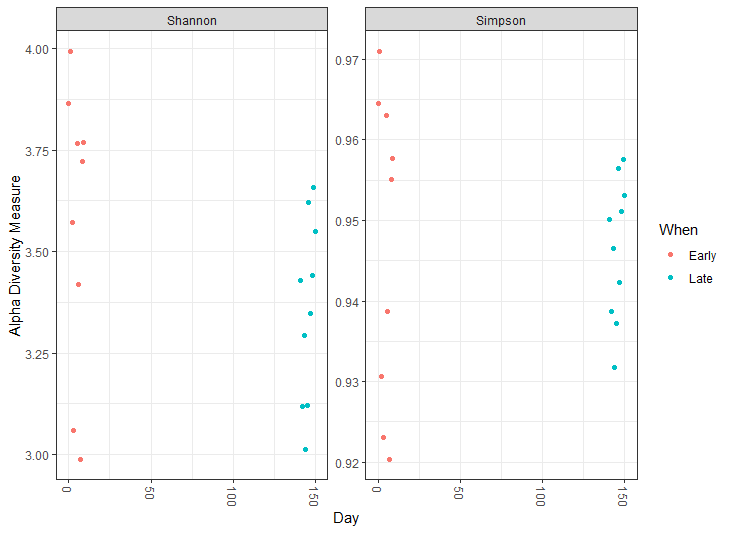
이렇게 하면 그림은 그려지지만 error 메시지가 뜨게 된다
Error in thene_bw() : could not find function "thene_bw"
In addition: Warning message:
In estimate_richness(physeq, split = TRUE, measures = measures) :
The data you have provided does not have any singletons. This is highly suspicious. Results of richness estimates (for example) are probably unreliable, or wrong, if you have already trimmed low-abundance taxa from the data.
We recommended that you find the un-trimmed data and retry.왜 이런 메세지가 뜨는가? data가 singletone을 가지고 있기 때문이다
그렇다면 singletone이란 무엇인가? ASV 긴 한데 모든 샘플 중 딱 하나만 가지고 있는 ASV를 말한다
그러나 singletone이 많으면 다양성이 높게 나온다. 즉 singletone을 시퀀싱 에러로 간주하여 삭제하는 것이 좋다
하지만 이 singletone이 시퀀싱 오류일 수도 있고 진짜 딱 하나의 샘플에서만 검출된 ASV일 수도 있다
2) Beta diversity Ordination and Plots
# Transform data to proportions as appropriate for Bray-Curtis distances
ps.prop <- transform_sample_counts(ps, function(otu) otu/sum(otu))
?ordinate
# "DCA", "CCA", "RDA", "CAP", "DPCoA", "NMDS", "MDS", "PCoA"
# "jsd" "dpcoa" "wunifrac" "unifrac" "bray"각 샘플마다 read의 총합이 다르기 때문에, 각 총합으로 각각 샘플에 포함된 ASV의 read count를 나누어 주어 normalization을 해준다. 이는 transform_sample_counts()로 수행 가능 하다
Beta diversity를 시각화하기에 앞서 각각의 데이터를 분류할 때 사용하는 값("jsd" "dpcoa" "wunifrac" "unifrac")과 분류방법("DCA", "CCA", "RDA", "CAP", "DPCoA", "NMDS", "MDS", "PCoA")을 선택해야 한다
보통 micobiome은 "wunifrac" "unifrac" "bray"와 "PCoA"를 많이 사용한다
자세한 개념은 여기에 다 적을 수 없음으로 다른 글에서 적어보겠다
bray matrix를 사용해 NMDS plot을 한번 그려보자
ord.nmds.bray <- ordinate(ps.prop, method="NMDS", distance="bray")
plot_ordination(ps.prop, ord.nmds.bray, color="When", title="Bray NMDS")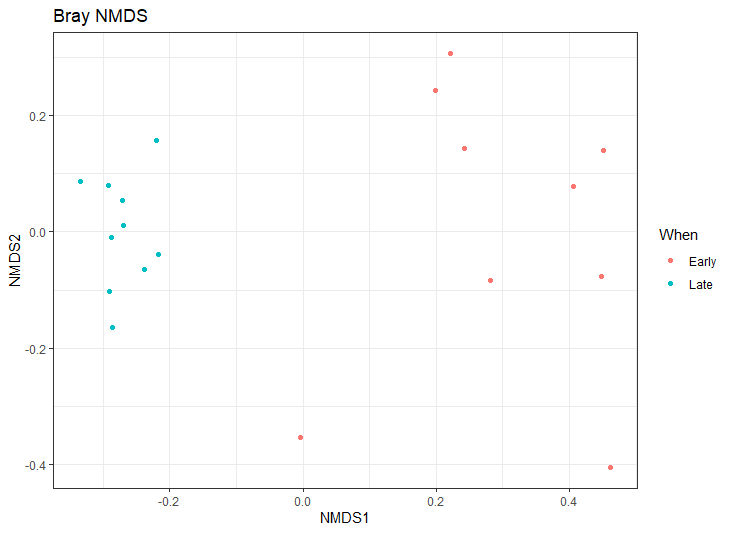
색은 When을 기준으로 표시하였는데, 각 그룹이 겹치지 않고 잘 나뉘는 것을 볼 수 있다
beta diversity plot을 해석하는 법은 각기 다른 처치를 한 그룹이 다르게 묶일수록 결과가 잘 나온 것이다.
ord.nmds.Uni <- ordinate(ps.prop, method="PCoA", distance="unifrac")
plot_ordination(ps.prop, ord.nmds.Uni, color="When", title="Unifrac PCoA")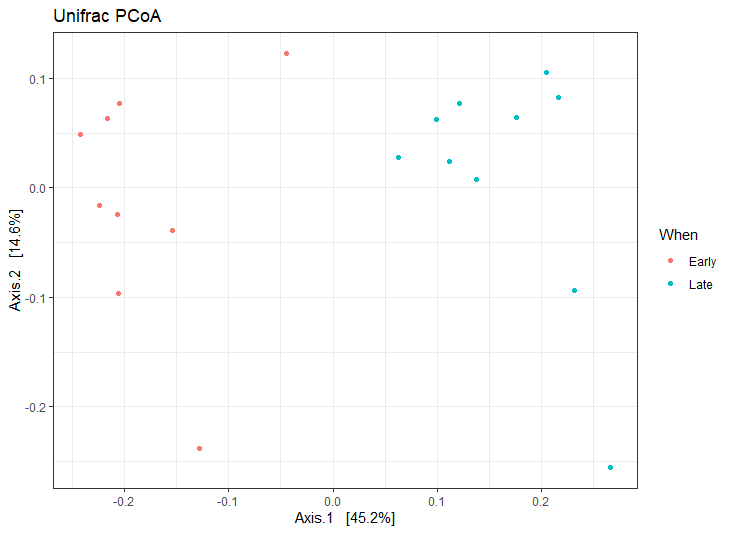
3) Phlogenic Tree
Top20개의 서열만 그려보자
top20 <- names(sort(taxa_sums(ps), decreasing=TRUE))[1:20]
ps.top20 <- transform_sample_counts(ps, function(OTU) OTU/sum(OTU))
ps.top20 <- prune_taxa(top20, ps.top20)
plot_bar(ps.top20, x="Day", fill="Family") + facet_wrap(~When, scales="free_x")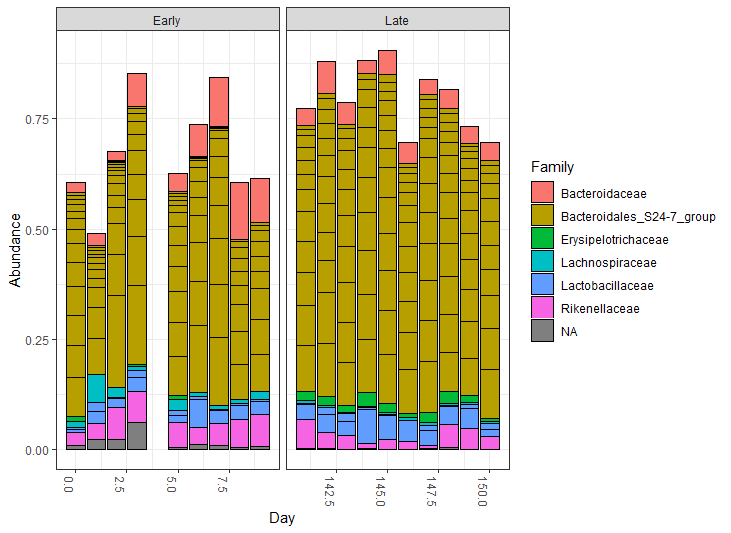
그 외에도 다양한 시각화 패키지가 존재함으로 차후에 다뤄볼 예정이다