
이전 편
📗인코렌탈(생물정보 분석): 언제 어디서나 클라우드를 이용한 NGS분석 01
1. 서버 접속 이후
서버 접속이 완료되었다면, 아래와 같은 화면이 정상적으로 보입니다.
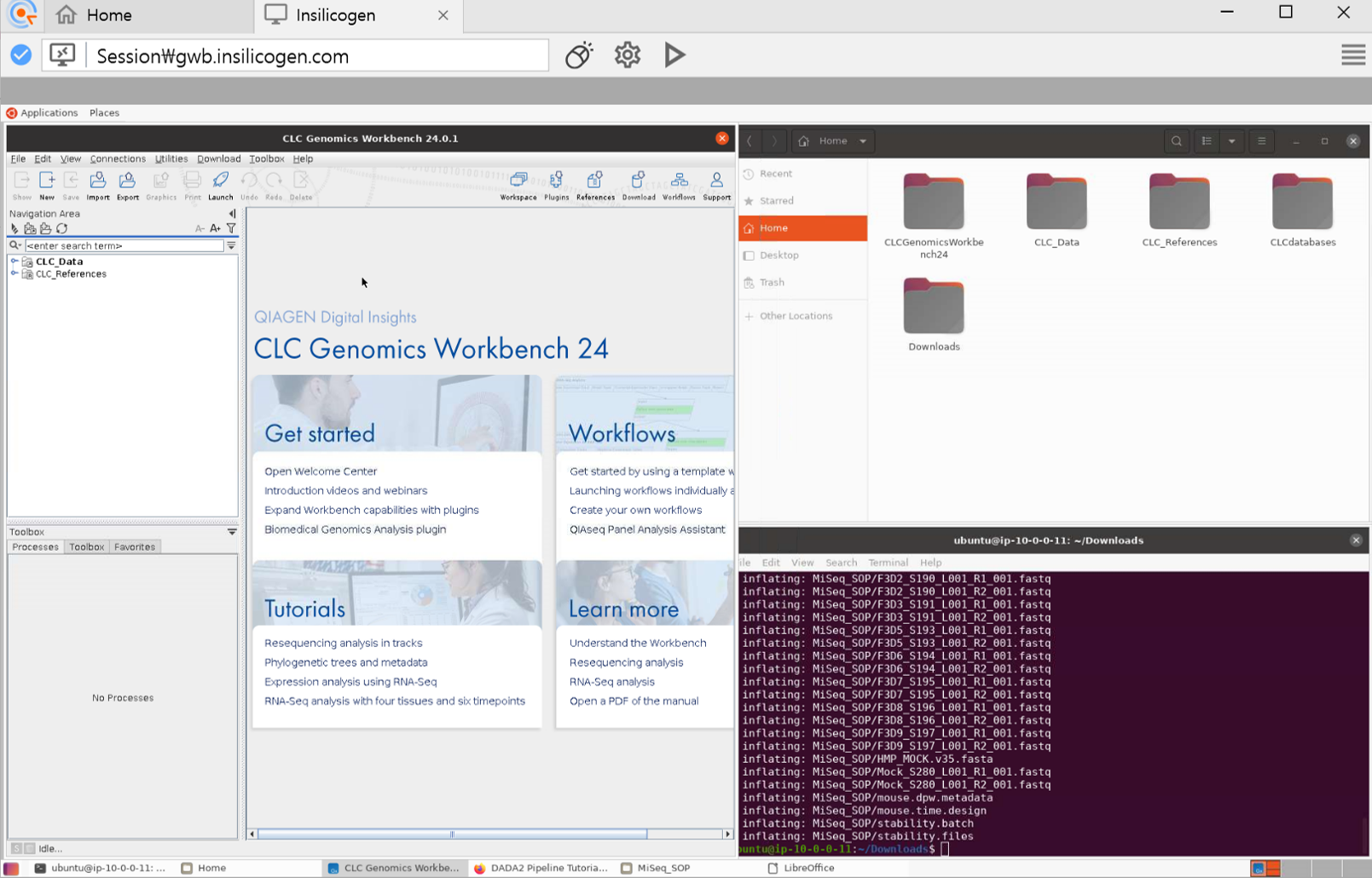
왼쪽에는 CLC Genomics Workbench 가 있으며, 우측 상단에는 리눅스 OS의 폴더와, 아래는 파일을 주고받기 위한 commend line 창이 띄워져 있습니다.
제가 사용한 파일은 엠플리콘 마이크로바이옴 예제 데이터입니다. Miseq으로 수행되었으며, 16S rRNA의 V4를 forward와 reverse방향에서 읽어낸 파일입니다(paired-end). 위 샘플은 DADA2의 예제 파일로서, 38개의 데이터로 구성되어 있습니다.
2. 지원하는 분석단계
1) 개별 도구
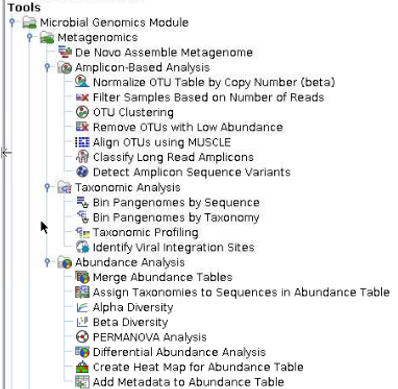
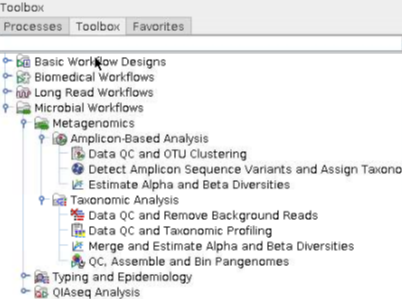
CLC의 왼쪽 아래 Tool box를 보면, 위 솔루션에서 지원하는 도구들이 기능에 맞게 트리구조로 정렬되어 있습니다.
미생물 유전체 분석, 특히 엠플리콘 마이크로바이옴 분석을 위해 위 사진에 있는 단계들을 지원합니다. 각 단계별로 클릭하여, 순서대로 진행이 가능합니다.
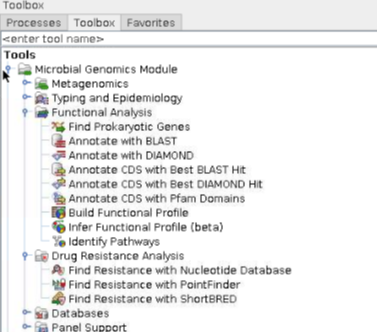
아래 추가적인 미생물 기능유전체분석과 항생제 저항성을 분석 도구도 지원하고 있습니다.
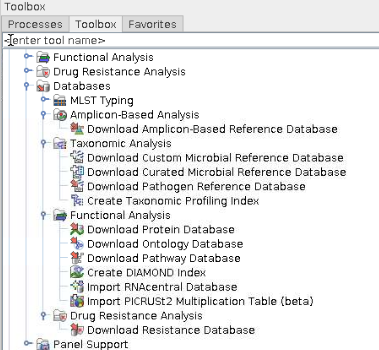
추가적으로 미생물 유전체 데이터 베이스 관련하여 Multilocus Sequence Typing(MLST) 절차도 제공하고 있습니다. 또한 Alignment를 위해 기존 오픈소스 데이터 베이스(Greengene, SILVA, UNITE 등)를 CLC에서 자유롭게 사용 가능합니다.
2) Workflow
혹은 각 단계를 통합한 workflow를 선택하여 수행할 수 있습니다.
3) Amplicon microbiome analysis
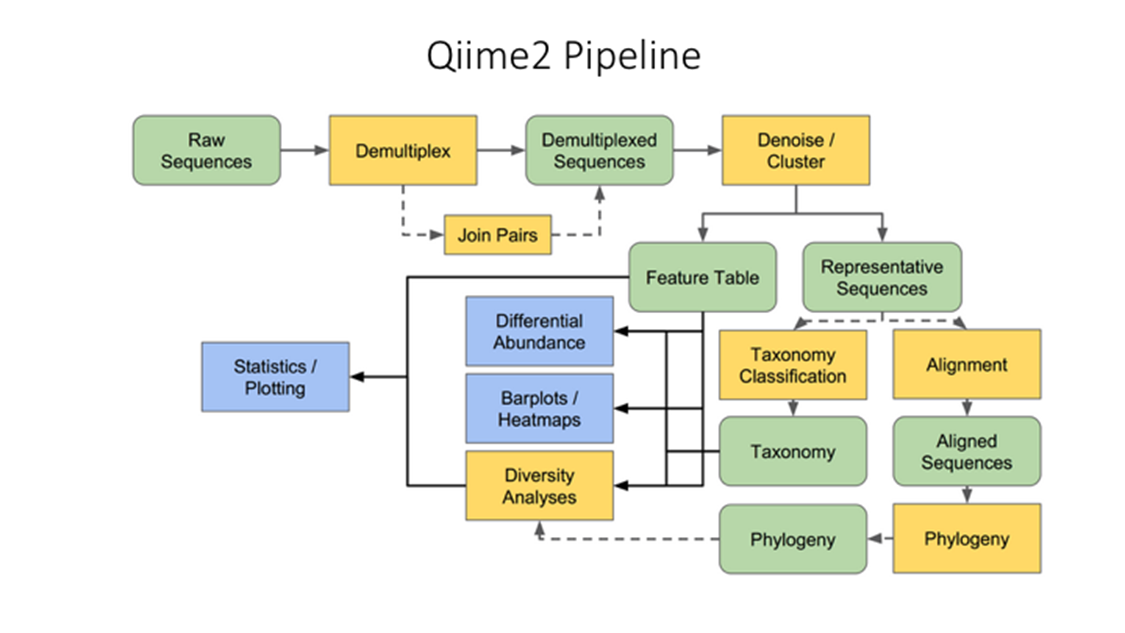
이번 글에서는 CLC Genomics Workbench Premium를 사용한 microbiome amplicon 분석을 수행해 보겠습니다!
paired-end 형식의 FASTQ 샘플을 ASV분석과 Taxnomy assignment이후, Diversity를 관찰하고 통계분석을 수행해 보겠습니다..
이때, Microbiome workflow를 사용해 보겠습니다.
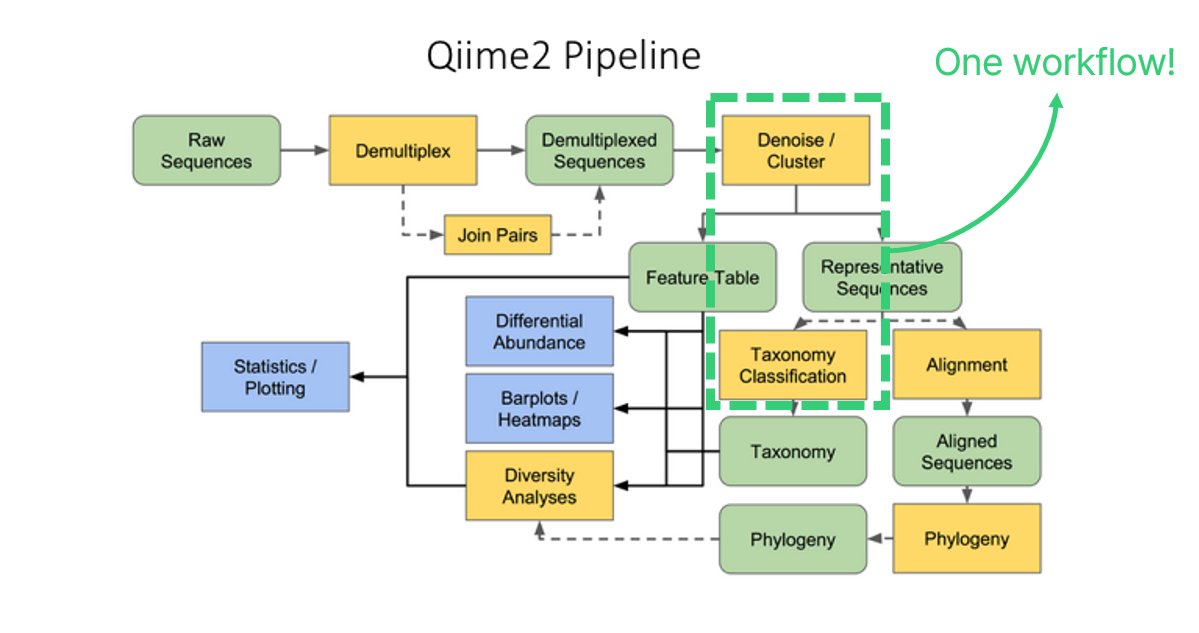
만약 "Detect Amplicon Sequence Variants and Assign Taxonomy" workflow를 사용하면, 아래 그림과 같은 두 단계를 하나의 분석으로 수행 가능합니다.
3. 파일 업로드
CLC Genomics Workbench Premium는 다양한 형식의 데이터를 지원합니다. 특히 NGSNGS 같은 경우 시퀀싱 플랫폼에 따라 파일형식이 다양합니다(HIFI, fastq, …). CLC는 NGS분석에 가장 많이 사용되는 일루미나 사의 Paired-end 시퀀싱 방식의 결과물을 당연히 지원합니다.
1) FASTQ 파일 불러오기
제가 사용하는 샘플은 Illumina사의 paired-end 방식으로 제작된 샘플이기 때문에, import형식에서 이를 설정해 줍니다.
(1) [File]-[Import]-[Illumina] 클릭
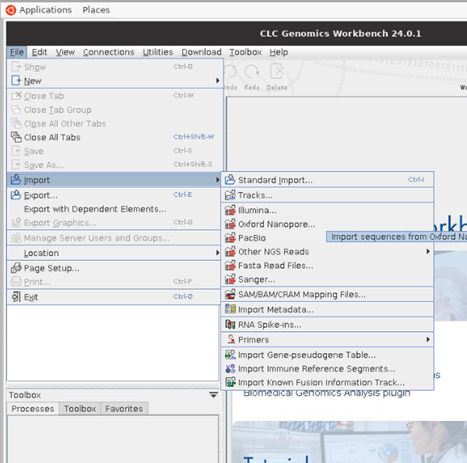
(2) [Add folder]로 샘플이 지정된 폴더를 선택하거나, [Add files]을 사용하여 개별 샘플 선택
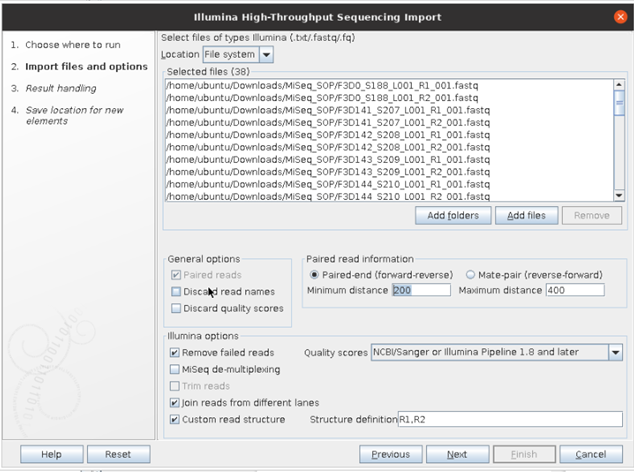
- Paired read information: Paired-end 형식에 필요한 추가 옵션 제공
- Illumina option: 일루미나 사의 데이터(특히 Miseq)에 대한 추가 기능 제공
- Structure definition: forward와 reverse서열의 이름이 어떻게 라벨링 되었는지 세부적으로 작성
2) Metadata 불러오기
샘플이 사람 샘플이라면, 이 샘플 대상자의 성별이 어떠한지, 질병상태인지 등을 기록한 메타데이터를 추가로 넣어주어야 합니다.
(1) Meta data를 excel 파일 혹은 csv 파일로 저장
(2) 솔루션 내에서 [File]-[Import]-[Import metadata] 클릭
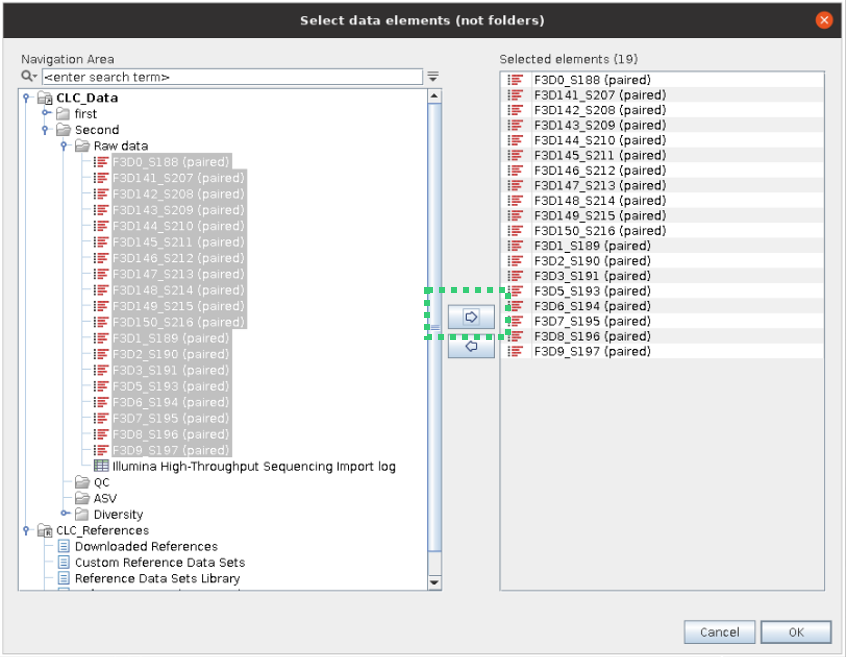
- 위 그림에서, Meta data와 연관된 파일을 오른쪽으로 넘겨줍니다.
- 파일을 선택할 때는 맨 위 파일을 클릭하고, Shit를 누른 상태에서 가장 아래 파일을 클릭, 여러 파일을 한 번에 선택 가능합니다.
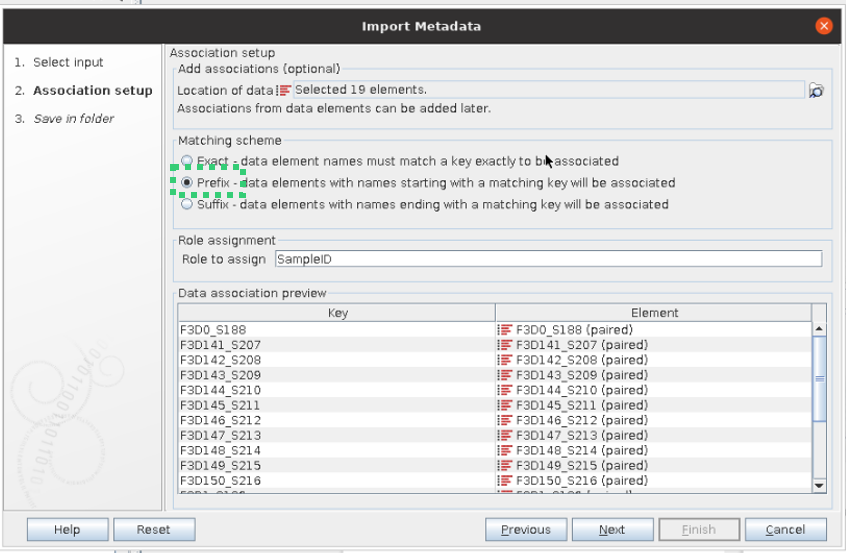
- 이후, 샘플 이름과 일치하는 데이터를 담고 있는 Metadata의 열이름 (e.g. SampleID)를 작성합니다.
- 메타데이터 구조 예시
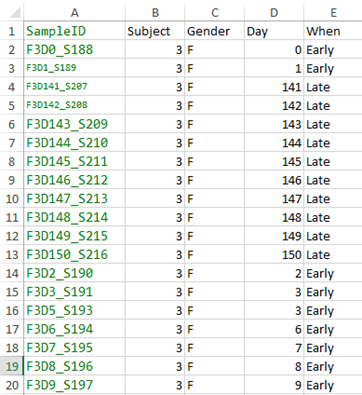
- 만약 38개 샘플이 3그룹으로 나누어져 있다면, 각 그룹별 추가 분석을 가능하게 합니다.
4. 샘플 필터링
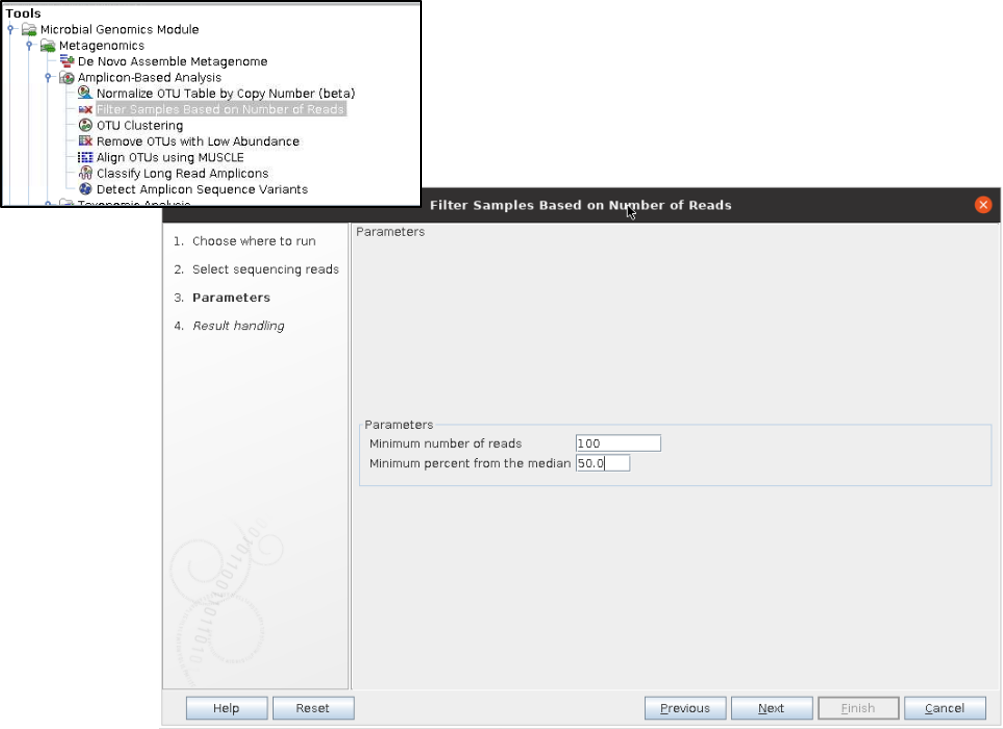
- 서열의 길이, 시퀀싱 depthdepth 혹은 퀄리티에 따라 샘플을 필터링해야 합니다.
- 위 예제 데이터는 Miseq을 이용해서 각 300bp씩 읽어낸 서열임으로, 길이가 100bp이하는 버려보도록 합시다.
- 이 솔루션의 장점은 Raw data값을 시각적으로 편리하게 확인 가능하다는 점입니다.
- 보통 Linux에서 확인하면 서열(ACCGGTTCC..) 값과(ACCGGTTCC..) 값과 Quality값이 ASCII code로만 표시되어 있지만, Workbench에서는 사람 친화적으로 확인이 가능합니다.
5. ASV 분석
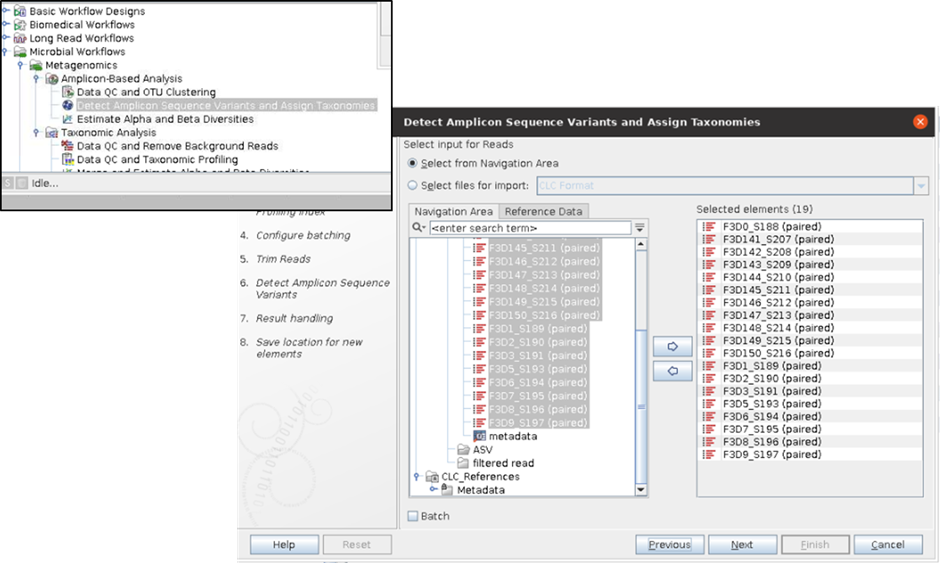
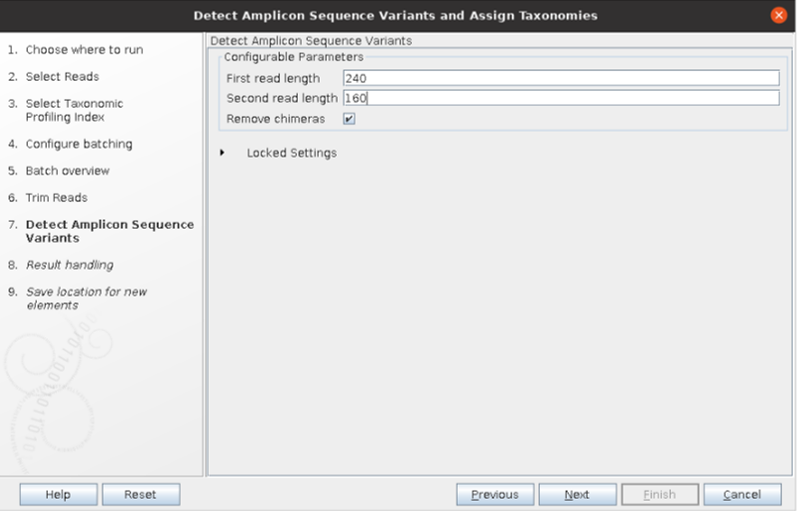
Workflow에 있는 Detect amplicon sequence variants and assign taxonomies를 선택합니다.
이는 ASV를 구하고, 여기에 바로 taxanomy정보를 추가하여 확인할 수 있습니다.
1) Workflow 수행
CLC에서 기본적으로 제공해 주는 SILVA SSU(small sub unit)을 사용해 봅시다.
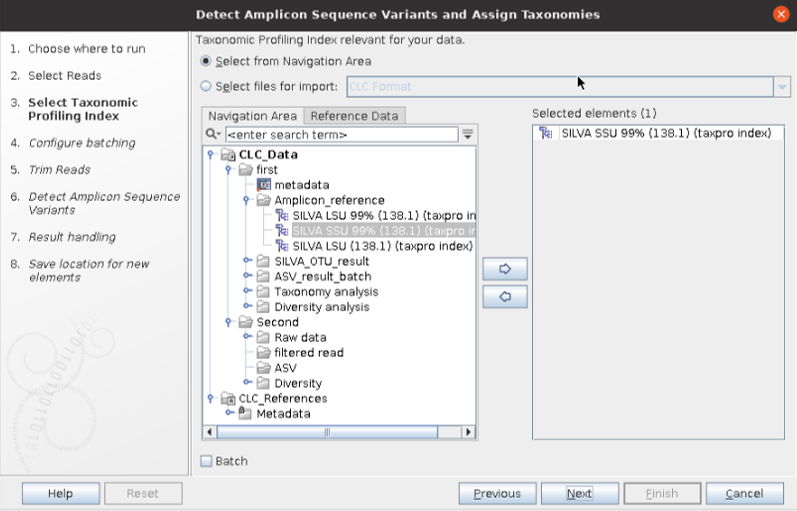
V4영역임으로, 퀄리티에 따라 forward는 250, reverse는 160으로 잘라주겠습니다.
결과적으로 개별 샘플에 따른 보고서와, 전체적인 보고서도 얻을 수 있습니다.
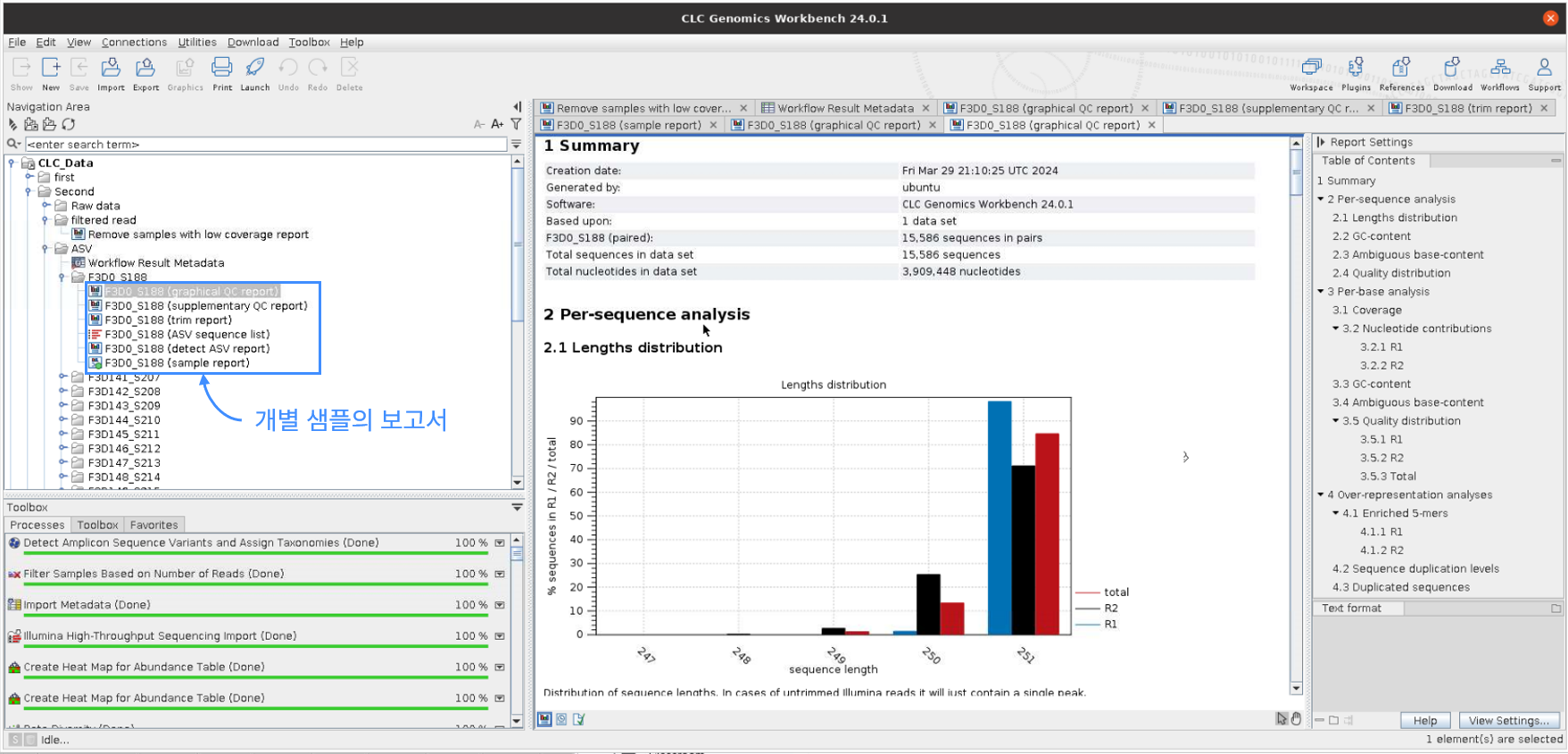
2) Report
(1) 개별 샘플 보고서
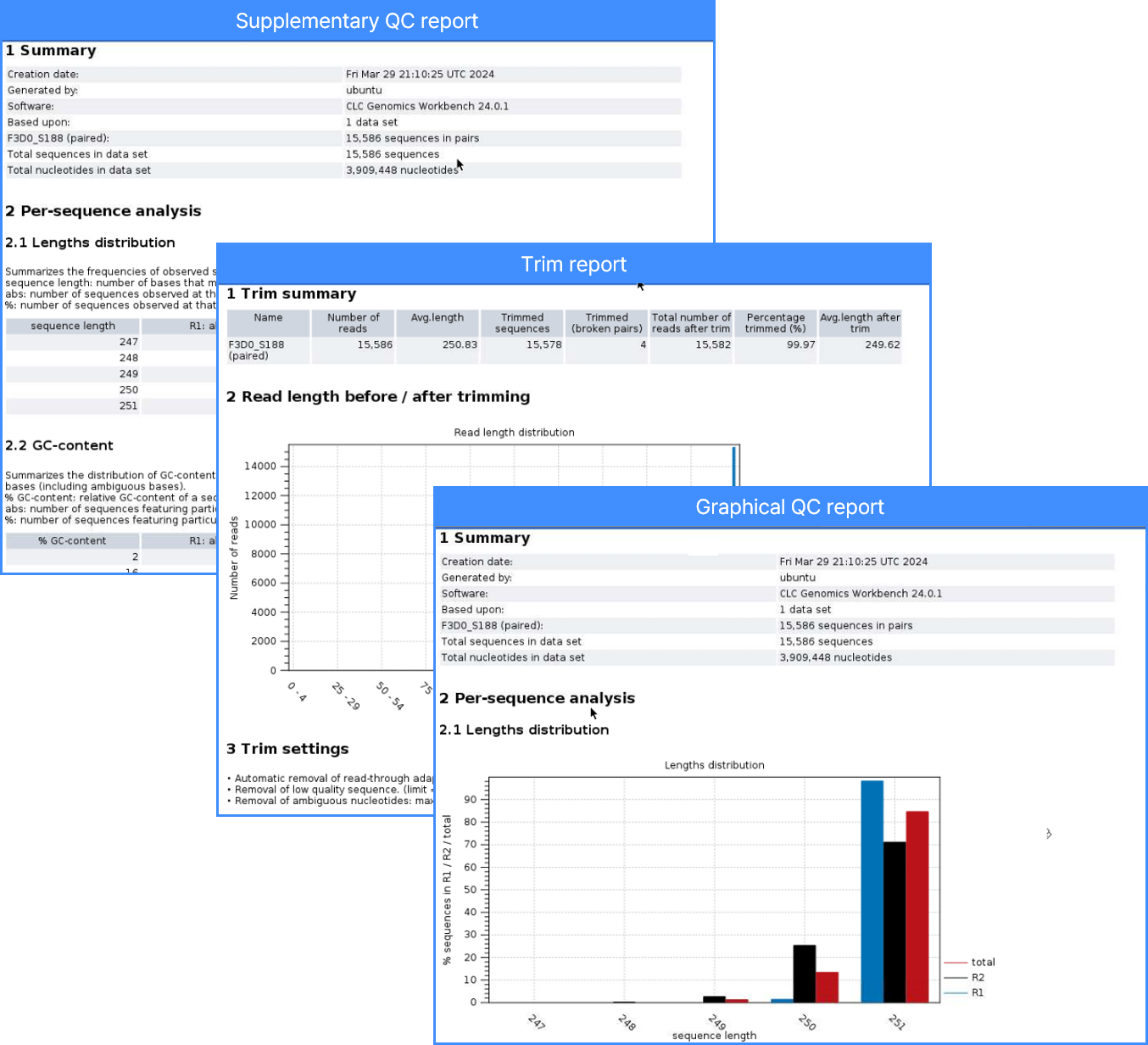
(2) 전체 샘플 보고서
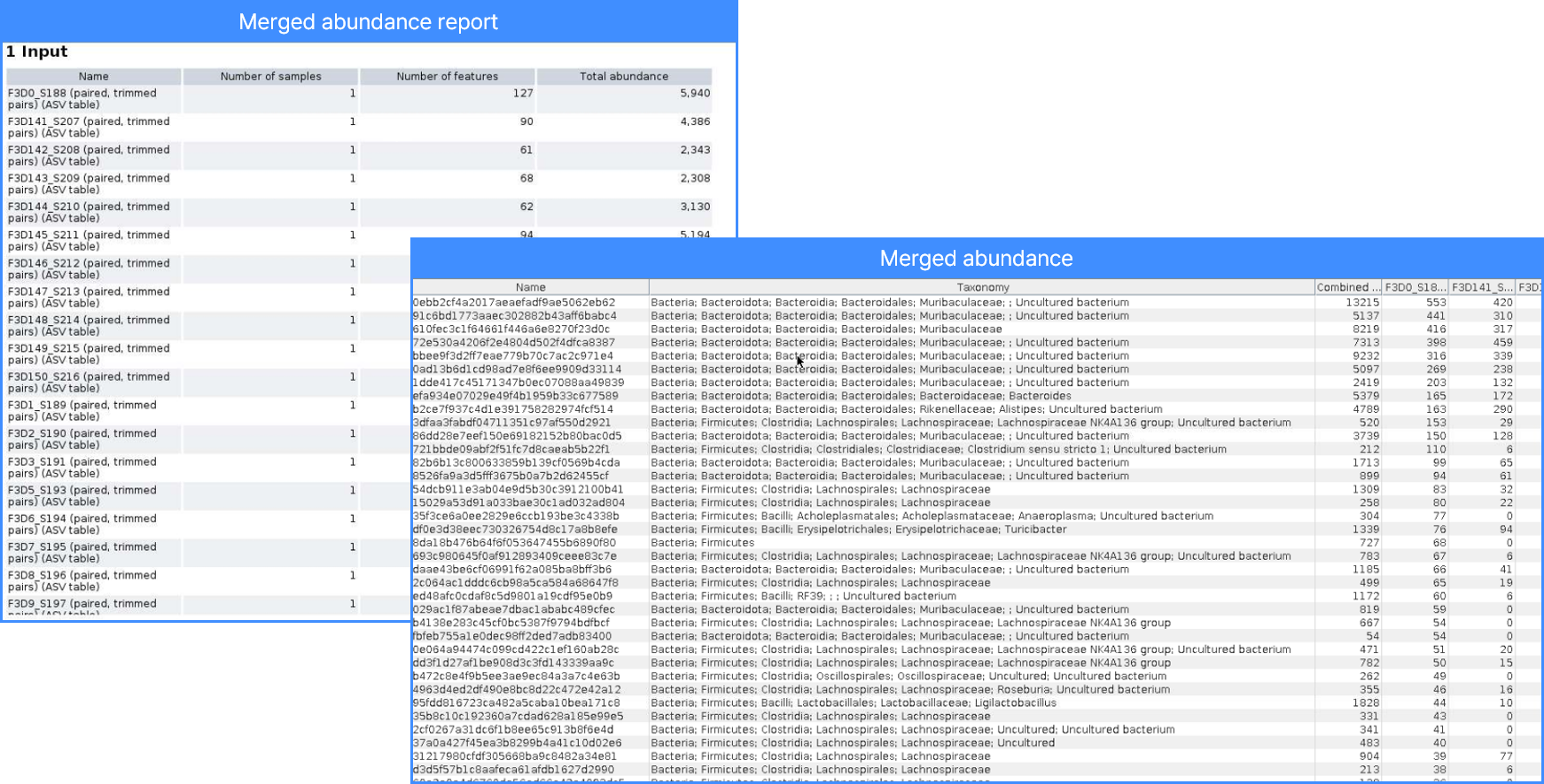
(3) ASV table과 taxonomy assignment 정보
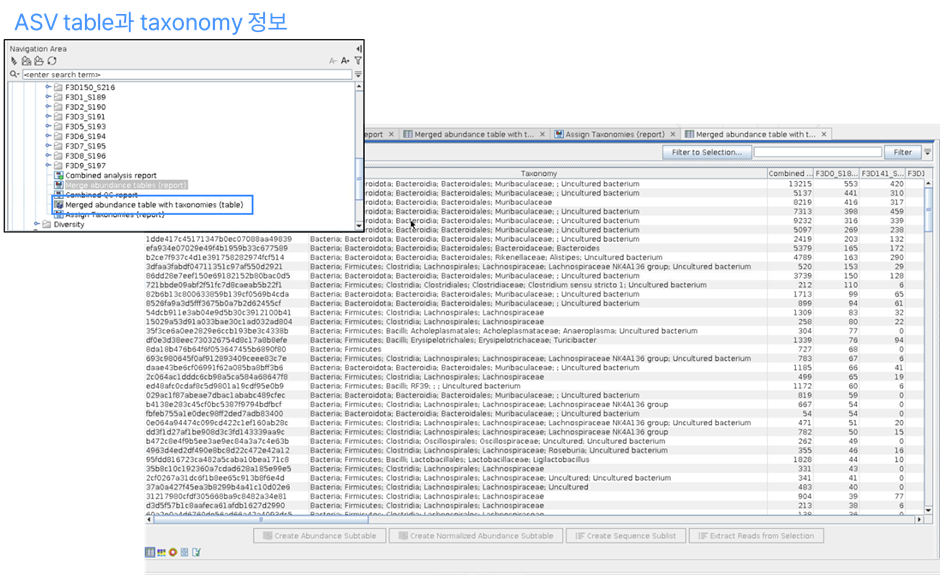
Merged abundance table with taxonomies를 보면, 전체 feature table과 각 ASV의 계통 정보를 확인할 수 있습니다.
6. 시각화하기
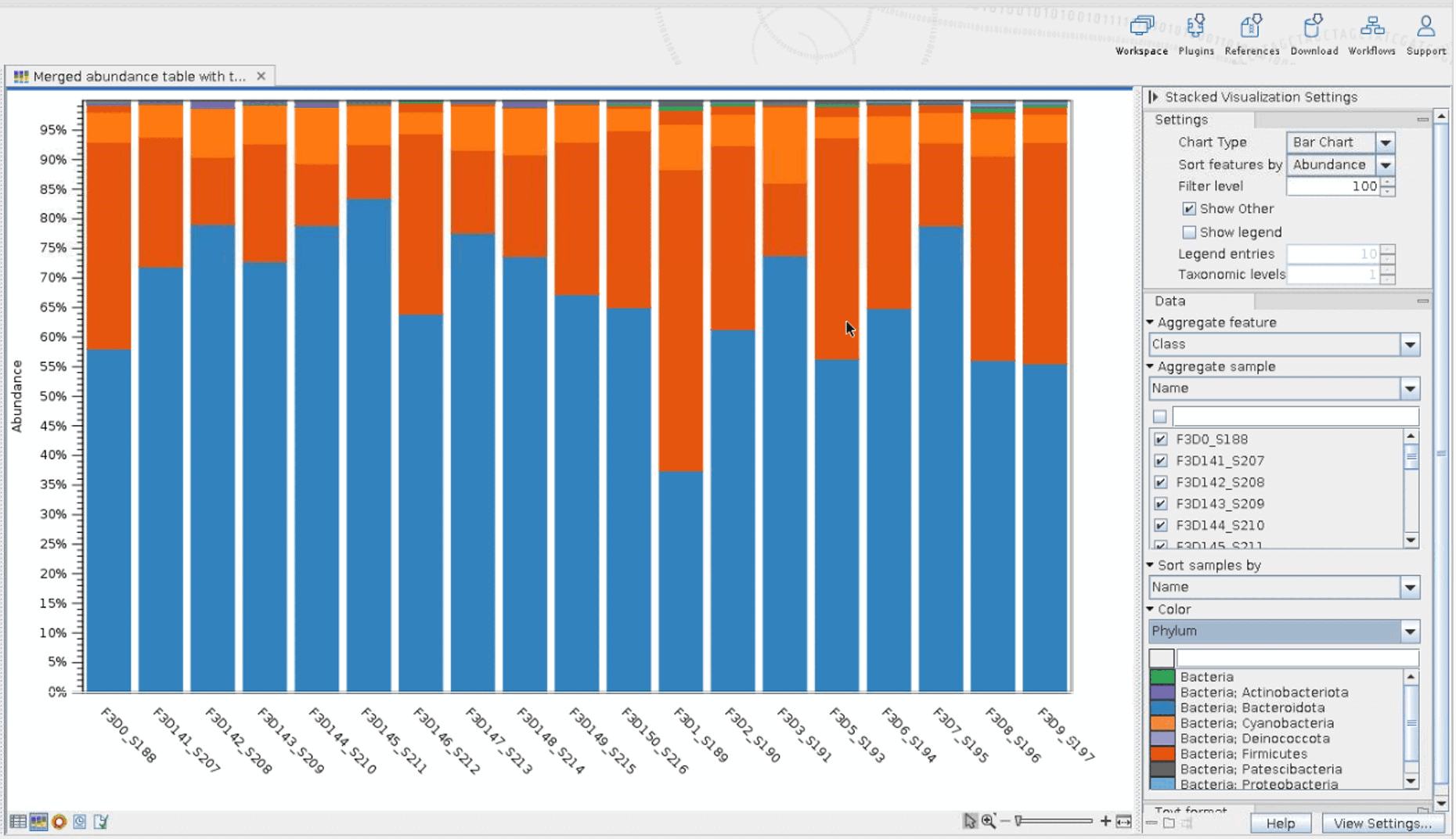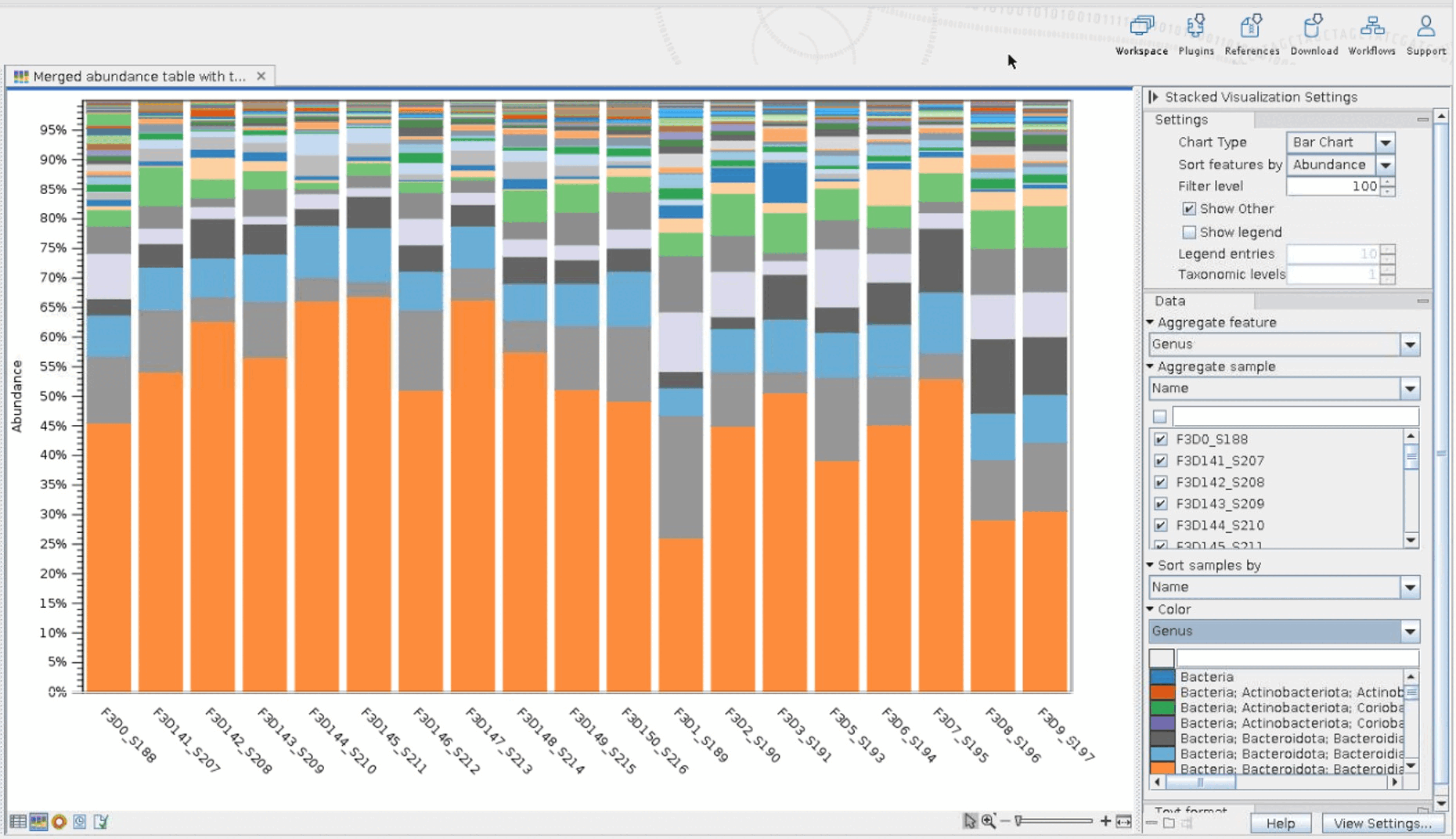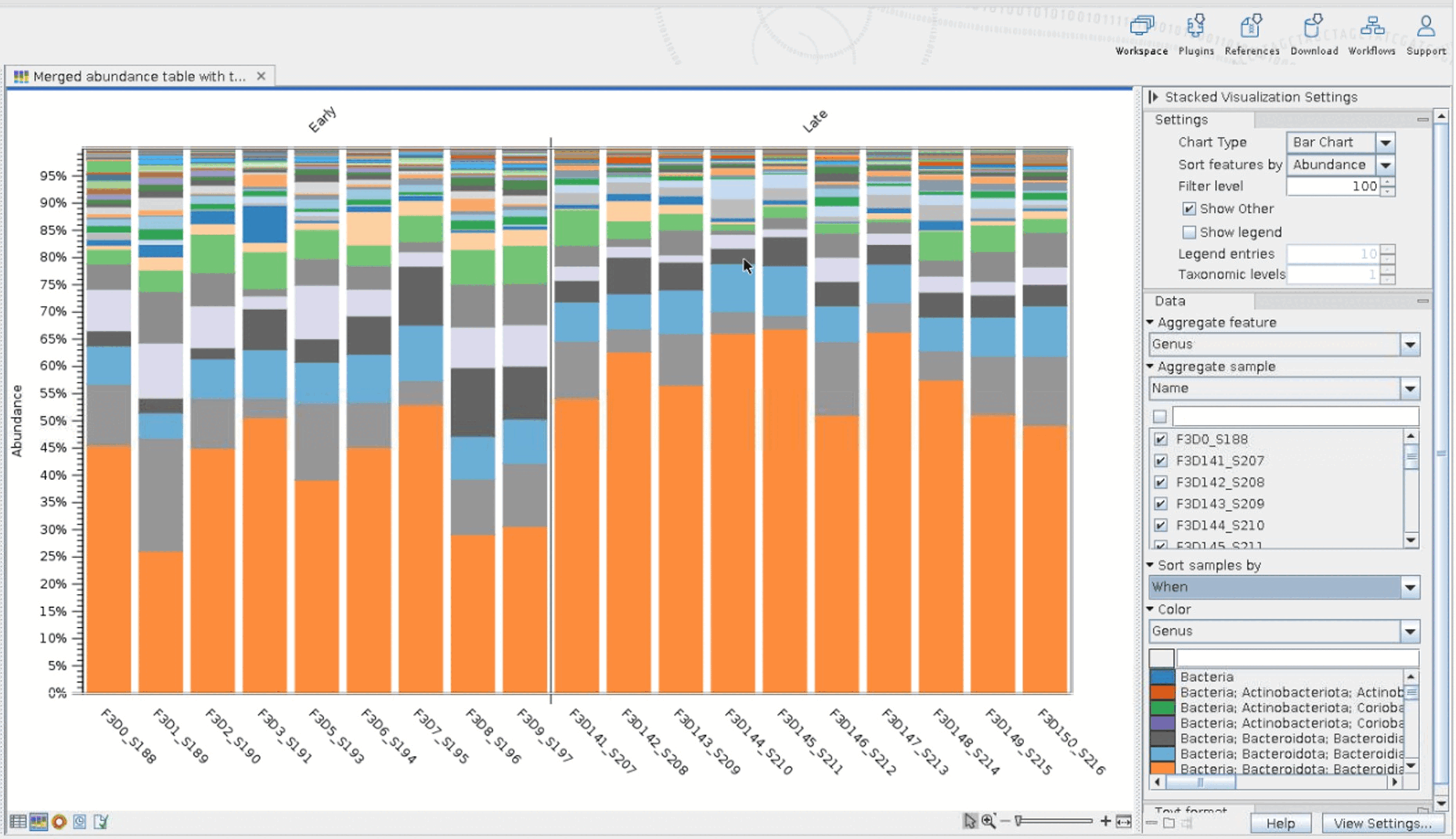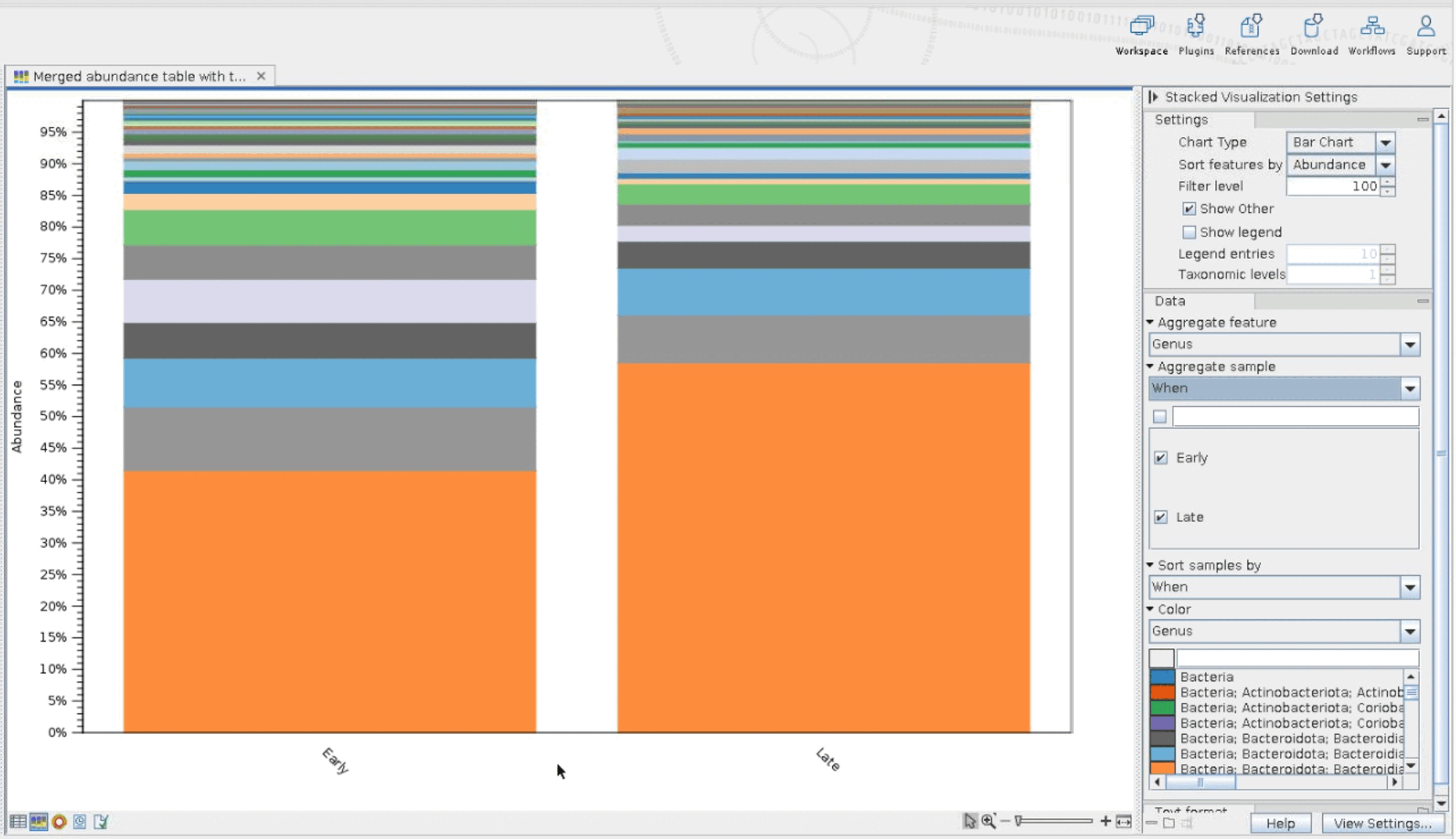
1) Taxonomy composition
Merged table에서 바로 bar plot으로 시각화하여,, 샘플의 계통적 다양성을 확인할 수 있습니다. Qiime View와 비슷하게 Genus, species level로 색을 달리하여 볼 수 있으며, 샘플이 아닌 각 그룹별로도 관찰이 가능합니다.
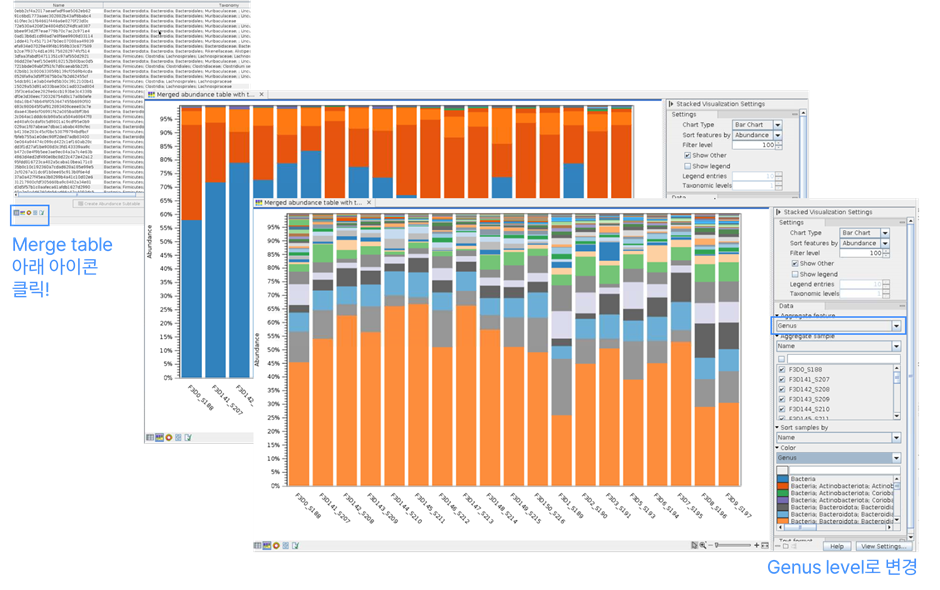
위 샘플은 Early와 late로 크게 두 그룹으로 나뉩니다. 이에 따라 왼쪽 옵션 창의 [Aggregate sample]을 metadata내의 해당 그룹이 속한 속성을 선택할 수 있습니다.
2) Diversity analysis
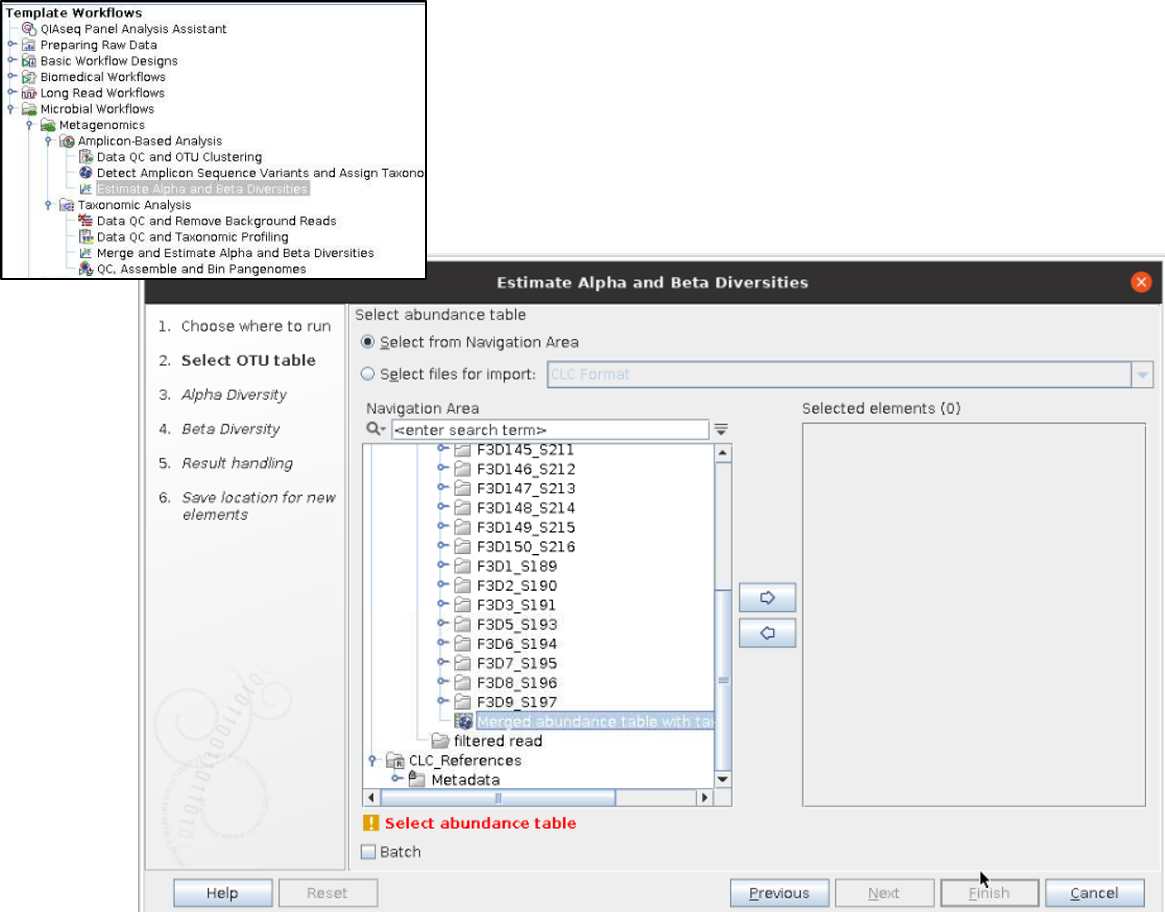
이제 alpha와 beta diversity를 분석해 봅시다!! Tool box에서 Estimate alpha and beta diversities를 사용해 한 번에 분석할 수 있습니다.
원하는 Alpha 지수와 와 Beta 거리를 선택한 후에, 각 값에 대한 결과와 시각화 값을 얻을 수 있습니다.
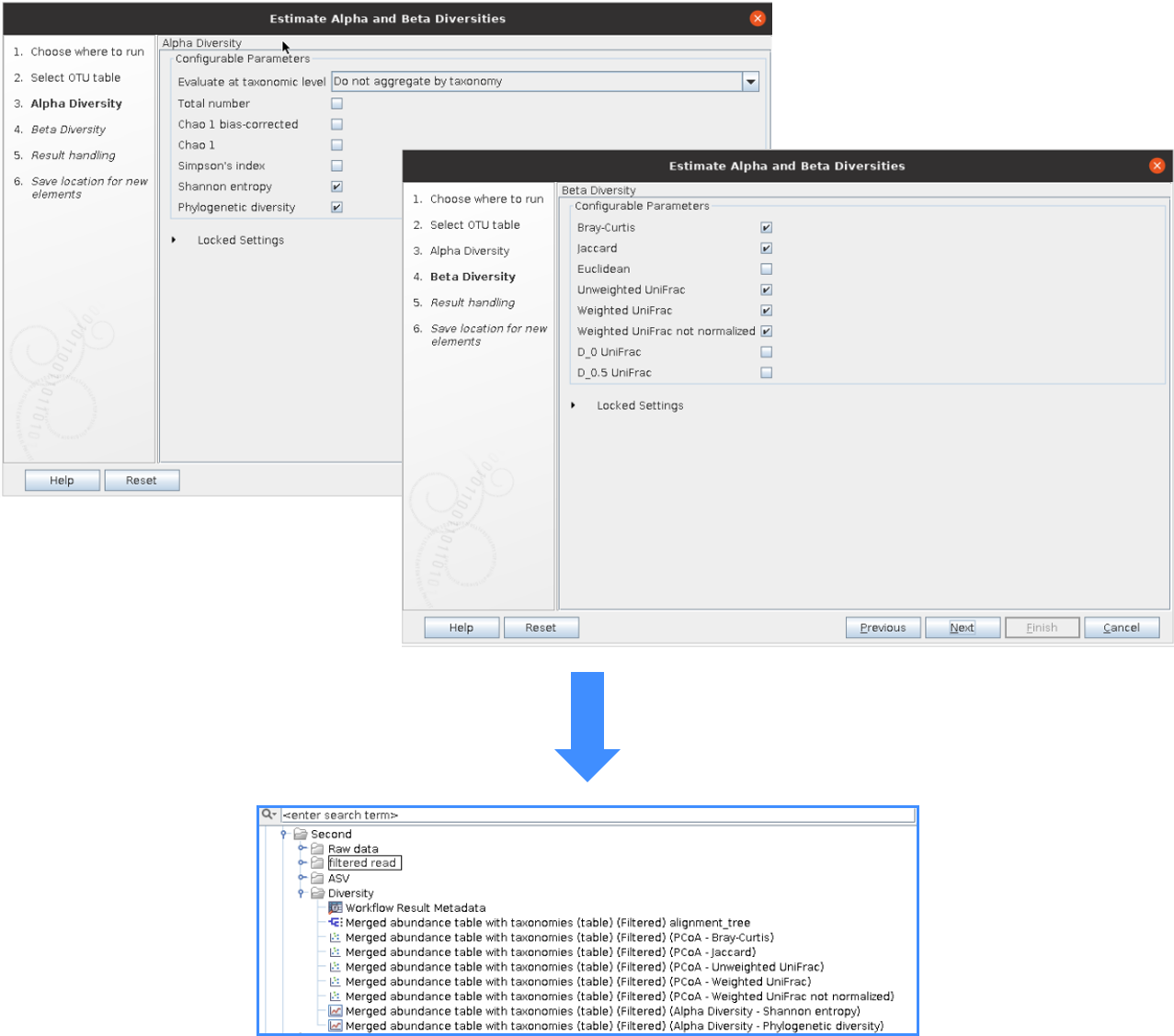
Unifrac 거리를 계산하기 위해선, 계통수가 만들어져야 합니다. 그러므로 위 workflow에서 모두 수행됩니다.
Alpha diversity부터 살펴봅시다. Alpha diversity 분석의 결과물의 아래 아이콘을 클릭하면, 바로 시각화가 가능합니다.
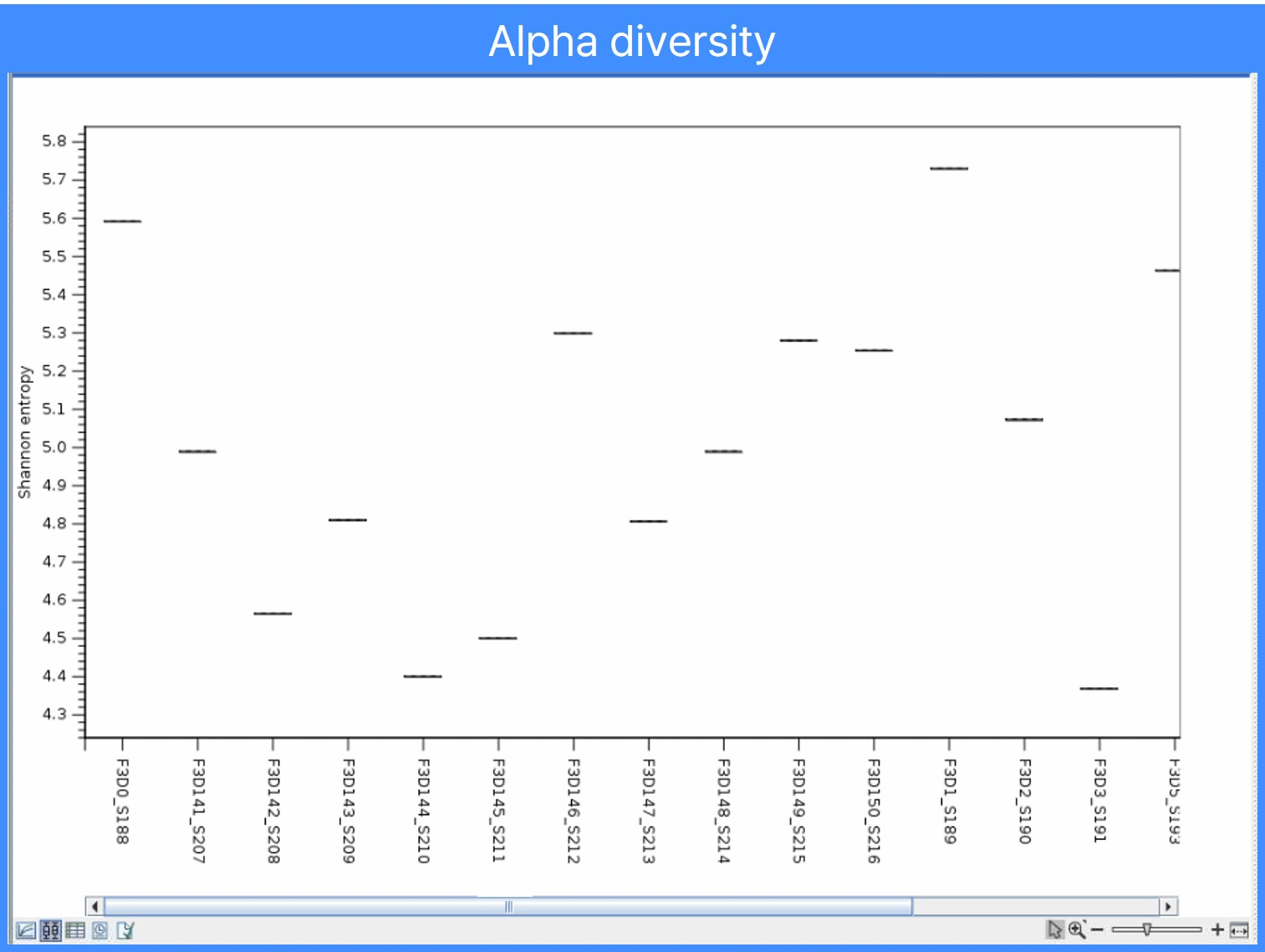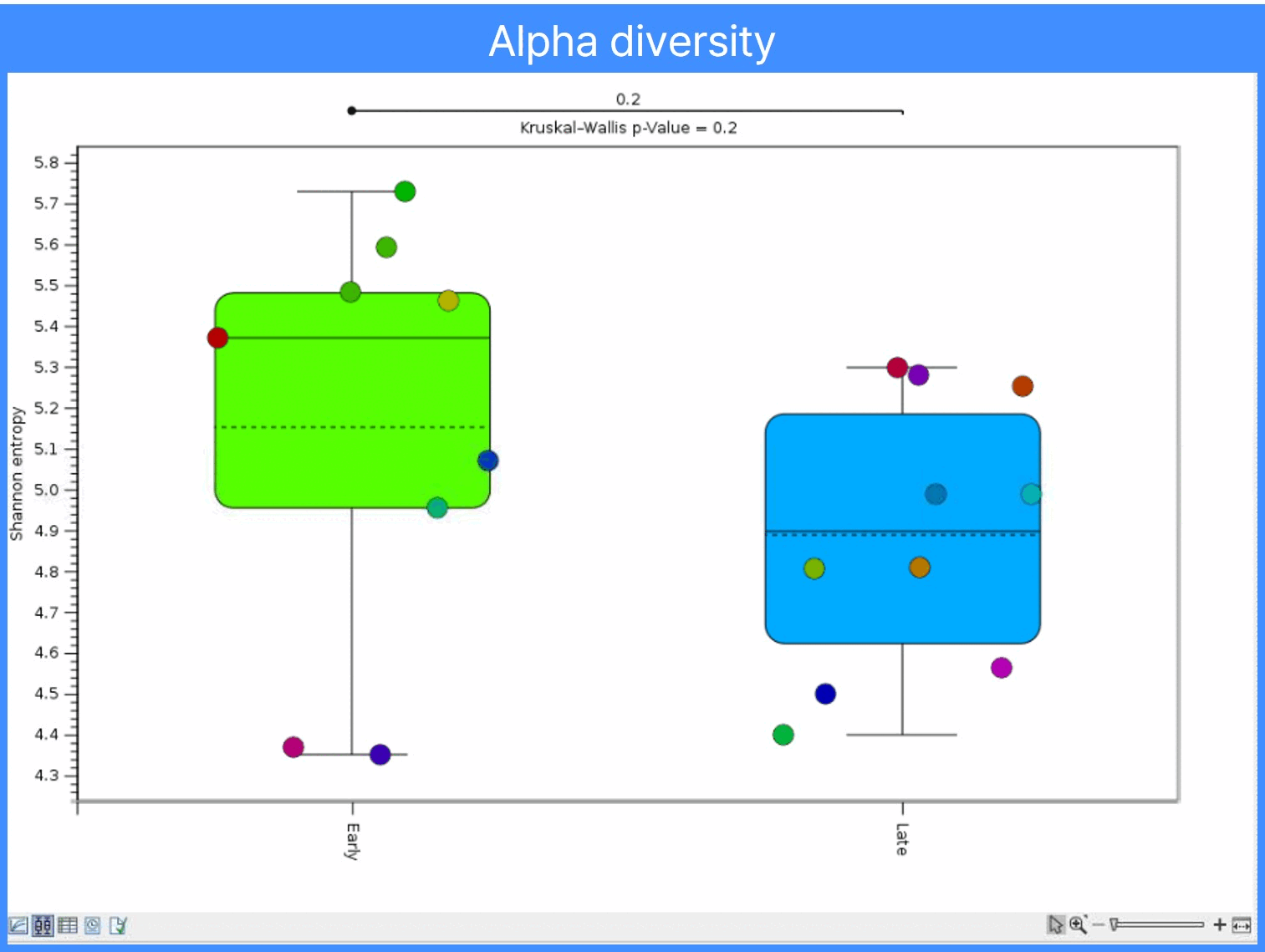
Bray-curtis 비유사도에 따른 Beta diversity분석 결과를 확인해 봅시다.. 이때 오른쪽 옵션값을 Group_by에서 우리가 비교하 하는 그룹을 넣습니다. 그러면, 각 샘플의 속성에 따라 색을 반영해 줍니다.
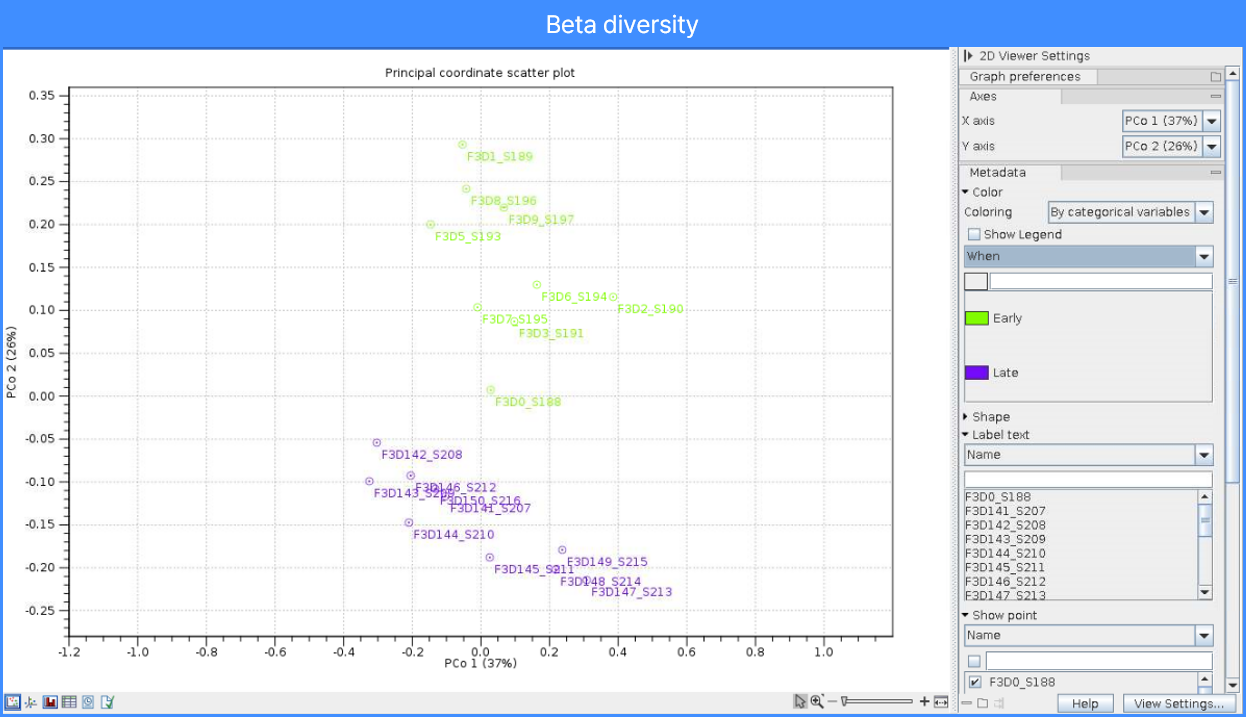
위 그림만 보아도 우리가 비교하고자 하는 그룹에 따라 미생물 분포가 확연하게 나뉘는 것을 볼 수 있습니다.
7. 통계분석
1) PERMANOVA
그러나 PCoA plot만 보고서는 각 그룹의 미생물 분포가 다른 지 짐작만 할 뿐입니다. 이를 통계학적으로 확증하는 단계가 필요합니다.
이를 위해 PERMANOVA 분석을 수행해 보겠습니다..
PERMONOVA란 PCoA plot상의 각 그룹의 중심점(centeroid)에서 각 샘플 간의 거리가 비교하는 그룹 간 차이가 있는지를 판별합니다.
이때, 비교하고자 하는 그룹을 선택합니다.
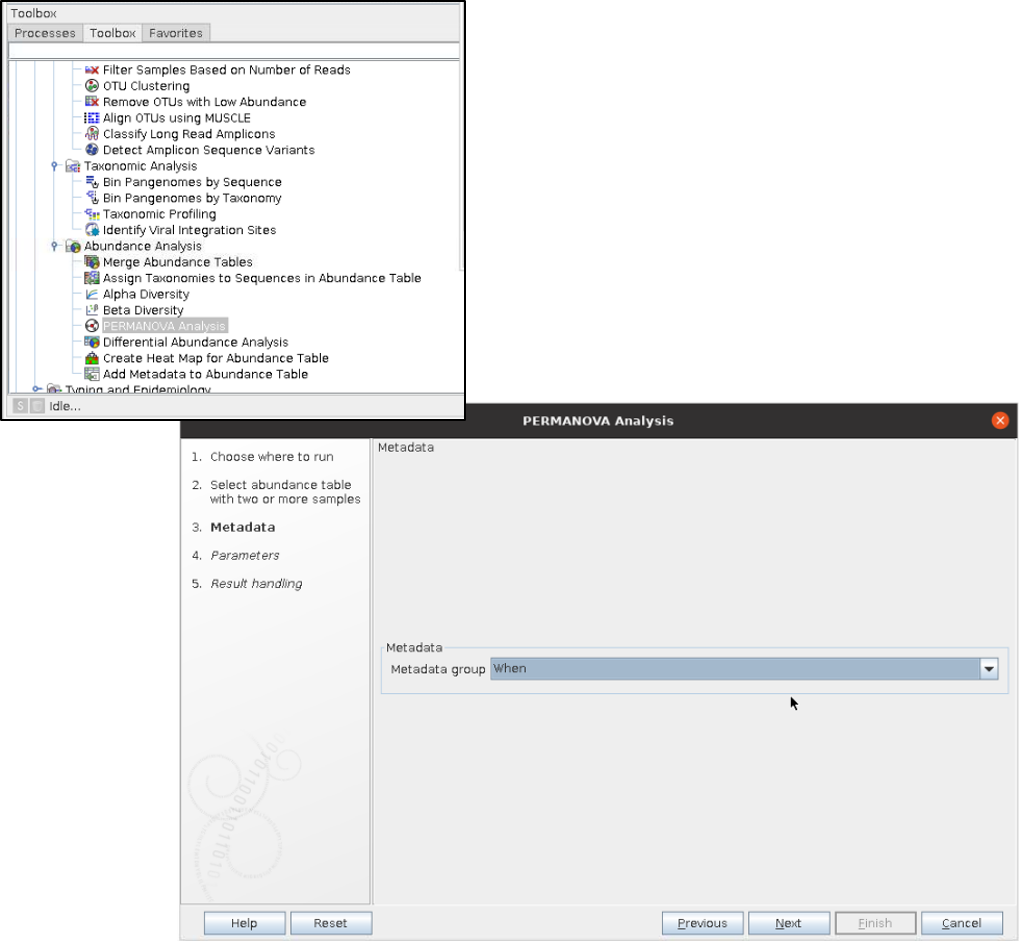
분석하고자 하는 거리를 선택합니다. 저는 Bray와 Jaccard를 선택하였습니다.
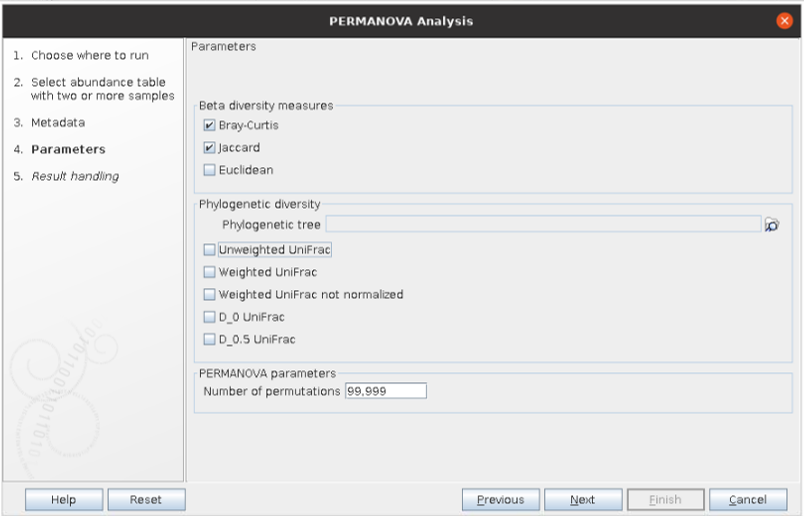
이후 PERMANOVA분석에 대한 보고서에서 p-value값을 확인하니 Bray와 Jaccard 거리에서 모두 매우 유의미한 것으로 나타났습니다(p-value < 0.001).

2) DAA
이게 각 그룹에만 특이적으로 풍부한 미생물이 어떤 것이 있는지 알아봅시다. 이러한 단계를 Differential abundance analysis(DAA)라고 합니다.
각 분석 단계에 대한 자세한 설명은 Help에서 찾아볼 수 있습니다.
DAA에 앞서, 우리는 계속 Early와 late그룹을 비교하였습니다.
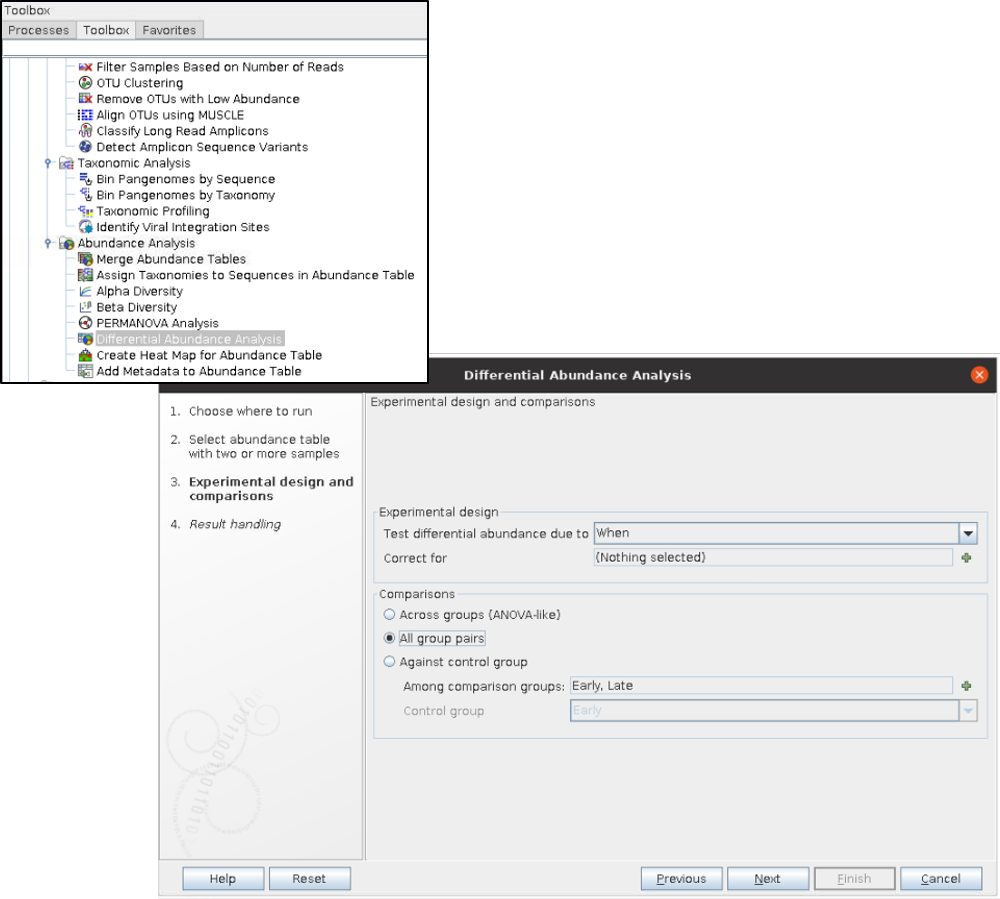
DAA분석에서도 두 그룹 간에 공통적인 ASV가 무엇인지, 혹은 각 그룹에만 존재하는 ASV가 무엇인지 알아봅시다.
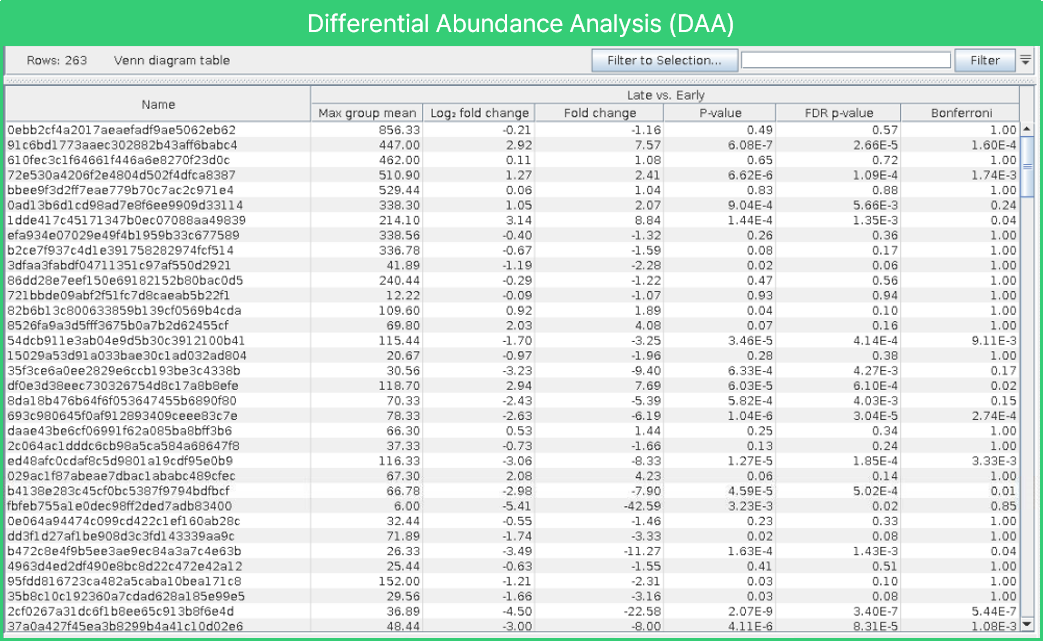
결과적으로 위와 같은 보고서가 얻어집니다. 이는 각 ASV가 그룹 간 차이가 유의한 지 판별합니다. Fold change로 양적 차이도 확인할 수 있습니다.
8. 사용후기
현재 저는 R기반의 DADA2를 사용하여 마이크로바이옴을 분석하고 있습니다. 마이크로바이옴 분석에서는 Qiime2가 가장 점유율이 높지만, 시각화 및 기타 분석을 모두 R에서 수행하기 때문에 저는 Linux보다는 R을 선호하는 편입니다.
하지만 DADA2라는 도구를 배우는 시간도 있지만, 결과물을 보여주기 위한 코드를 배우는 시간이 더 오래 걸렸던 것 같습니다. 프로그램 설치하면서 오류도 많이 겪고, 시각화를 위해 코드 오류를 일주일간 고쳤던 경험도 있네요.😅. 여하튼 아직도 코딩은 어렵습니다.
제가 직장인 분들과 마이크로바이옴, 메타게놈에 대한 교육을 받았던 기억이 있습니다. 대부분의 생물정보학 분석 교육은 기본적인 원리와 포괄적인 파이프라인에 대한 설명이 주를 이룹니다. 또한 알려준 코드는 예제 데이터에 맞추어져 있으므로, 바로 연구자의 데이터에 활용하기에는 분명히 한계가 존재합니다.
같이 교육을 들었던 직장인 분들은 실질적으로 우리가 직장에서 어떻게 적용하는지에 대해 알려주면 좋겠다고 하셨습니다. 즉, 아무리 분석의 원리와 단계를 잘 알아도 바로 실행 가능한 방법이 필요한 것입니다.
아마 이와 가장 가까운 방법이 인코렌탈이라는 생각이 듭니다. 인코렌탈을 그 과정(코드 학습 및 버전업데이트, 소프트웨어 설치 등등)에서 소요되는 시간낭비를 줄이고 효율적으로 오픈소스보다 더 나은 도구를 제공합니다. 또한 R기반 파이프라인과 비교하였을 때, 같은 데이터의 처리를 3시간에서 30분으로 줄여준다고 생각합니다.
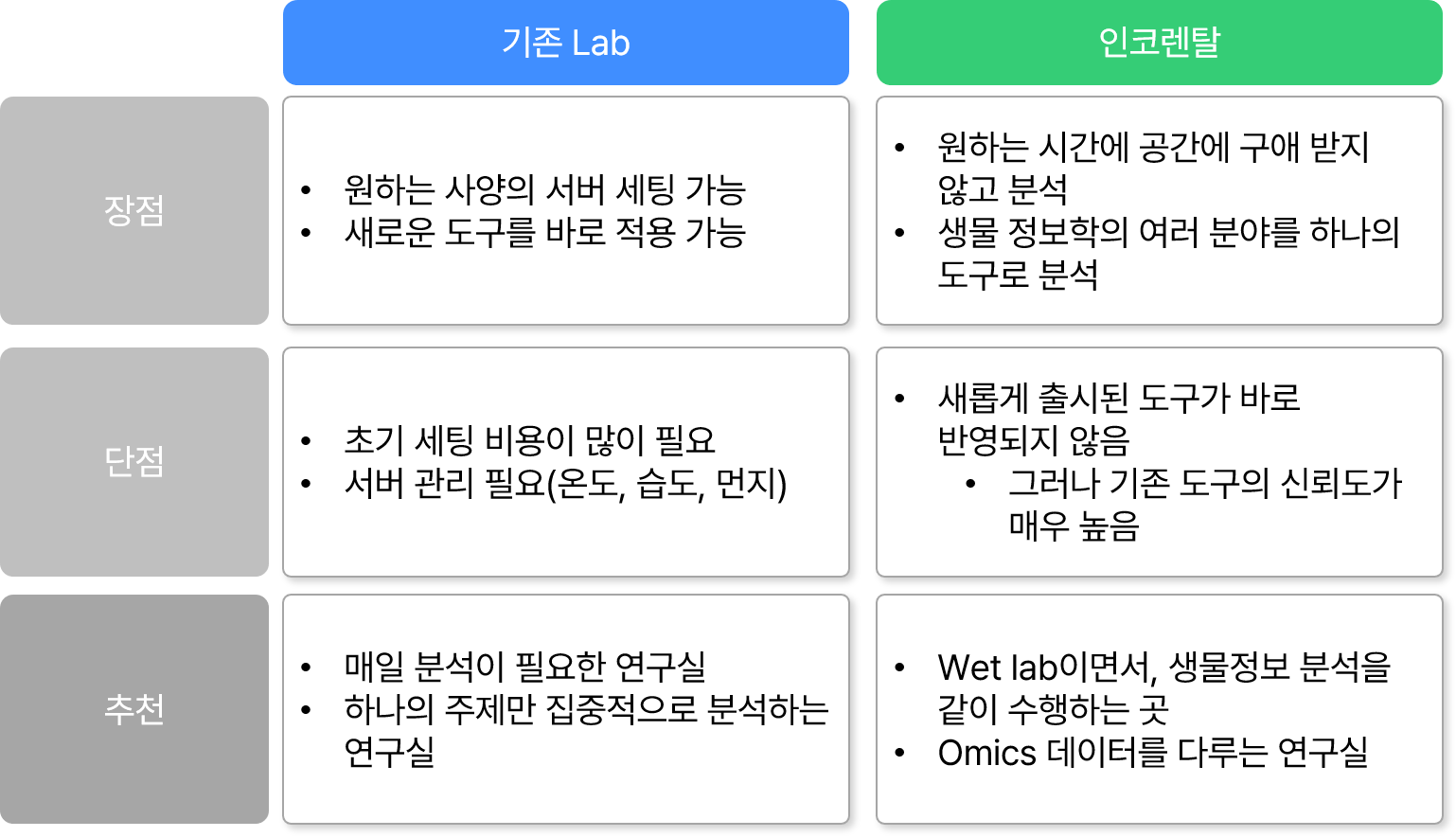
물론 새로운 도구를 바로 적용할 수 없다는 것이 CLC의 유일한 단점이라고 할 수 있습니다. 그러나 기존 도구들이 인용수가 높고, 신뢰도가 높은 도구만 사용하여 분석의 퀄리티와 결과는 전혀 뒤처지지 않는다고 생각합니다.
또한 생물정보학에서 Single cell이나 Transcriptomic 분석을 위한 방법을 배울 때, 하나의 분야만 배우는 대에도 정말 긴 시간이 걸립니다. 그러나 인코렌탈에 포함된 도구들은 이를 하나의 Workflow로 만들어 놓고, 클릭 한 번으로 실행 가능하게 합니다.
여러 연구에서 결국 중요한 것은 방법론이 아닌 연구의 결과라고 생각합니다. 인코렌탈 서비스는 부차적인 방법론에 대한 고민을 줄이고, 유의미한 연구 디자인과 결과 해석에 집중할 수 있게 하는 서비스라고 생각합니다.

해당 포스팅은 업체로부터 제품과 원고료를 지원받아 실제 사용한 후기를 작성하였습니다.