

🟦 BLAST 옵션의 단점
Local BLAST의 결과에서 우리가 확인할 수 있는 정보는 Tax id와 Phylum, Species이름 정보 등이다. 그러나 전체 계통 정보를 얻는 옵션은 기본적으로 추가되어있지 않다.
나는 기본적으로 blast 수행 시 아래와 같은 output format옵션을 선택한다.
-outfmt "7 delim=, qacc sacc evalue bitscore qcovus pident sscinames"결과는 아래와 같이
"query/ 접근번호/E-value/bitscore/query coverage/ identity/ 종속명"을 나타냅니다.

>> outfmt의 옵션 전체 보기
| 약어 | 내용 |
| qseqid | Query Seq-id (쿼리 시퀀스 ID) |
| qgi | Query GI (쿼리 GI) |
| qacc | Query accession (쿼리 액세션) |
| qaccver | Query accession.version (쿼리 액세션.버전) |
| qlen | Query sequence length (쿼리 시퀀스 길이) |
| sseqid | Subject Seq-id (대상 시퀀스 ID) |
| sallseqid | All subject Seq-id(s), separated by a ';' (모든 대상 시퀀스 ID, ';'로 구분) |
| sgi | Subject GI (대상 GI) |
| sallgi | All subject GIs (모든 대상 GI) |
| sacc | Subject accession (대상 액세션) |
| saccver | Subject accession.version (대상 액세션.버전) |
| sallacc | All subject accessions (모든 대상 액세션) |
| slen | Subject sequence length (대상 시퀀스 길이) |
| qstart | Start of alignment in query (쿼리에서 정렬 시작 위치) |
| qend | End of alignment in query (쿼리에서 정렬 끝 위치) |
| sstart | Start of alignment in subject (대상에서 정렬 시작 위치) |
| send | End of alignment in subject (대상에서 정렬 끝 위치) |
| qseq | Aligned part of query sequence (정렬된 쿼리 시퀀스 부분) |
| sseq | Aligned part of subject sequence (정렬된 대상 시퀀스 부분) |
| evalue | Expect value (기대값) |
| bitscore | Bit score (비트 점수) |
| score | Raw score (원시 점수) |
| length | Alignment length (정렬 길이) |
| pident | Percentage of identical matches (동일 일치 비율) |
| nident | Number of identical matches (동일 일치 수) |
| mismatch | Number of mismatches (불일치 수) |
| positive | Number of positive-scoring matches (긍정 점수 일치 수) |
| gapopen | Number of gap openings (갭 열기 수) |
| gaps | Total number of gaps (총 갭 수) |
| ppos | Percentage of positive-scoring matches (긍정 점수 일치 비율) |
| frames | Query and subject frames separated by a '/' (쿼리 및 대상 프레임, '/'로 구분) |
| qframe | Query frame (쿼리 프레임) |
| sframe | Subject frame (대상 프레임) |
| btop | Blast traceback operations (BTOP) (BLAST 추적 작업) |
| staxid | Subject Taxonomy ID (대상 계통학적 ID) |
| ssciname | Subject Scientific Name (대상 학명) |
| scomname | Subject Common Name (대상 일반명) |
| sblastname | Subject Blast Name (대상 BLAST 이름) |
| sskingdom | Subject Super Kingdom (대상 초왕국) |
| staxids | Unique Subject Taxonomy ID(s), separated by a ';' (고유 대상 계통학적 ID, ';'로 구분) |
| sscinames | Unique Subject Scientific Name(s), separated by a ';' (고유 대상 학명, ';'로 구분) |
| scomnames | Unique Subject Common Name(s), separated by a ';' (고유 대상 일반명, ';'로 구분) |
| sblastnames | Unique Subject Blast Name(s), separated by a ';' (고유 대상 BLAST 이름, ';'로 구분) |
| sskingdoms | Unique Subject Super Kingdom(s), separated by a ';' (고유 대상 초왕국, ';'로 구분) |
| stitle | Subject Title (대상 제목) |
| salltitles | All Subject Title(s), separated by a '<>' (모든 대상 제목, '<>'로 구분) |
| sstrand | Subject Strand (대상 스트랜드) |
| qcovs | Query Coverage Per Subject (대상별 쿼리 커버리지) |
| qcovhsp | Query Coverage Per HSP (HSP별 쿼리 커버리지) |
| qcovus | Query Coverage Per Unique Subject (고유 대상별 쿼리 커버리지, blastn 전용) |
이를 해결하고자 검색해 본 결과 아래와 같은 문의글을 찾을 수 있었다.
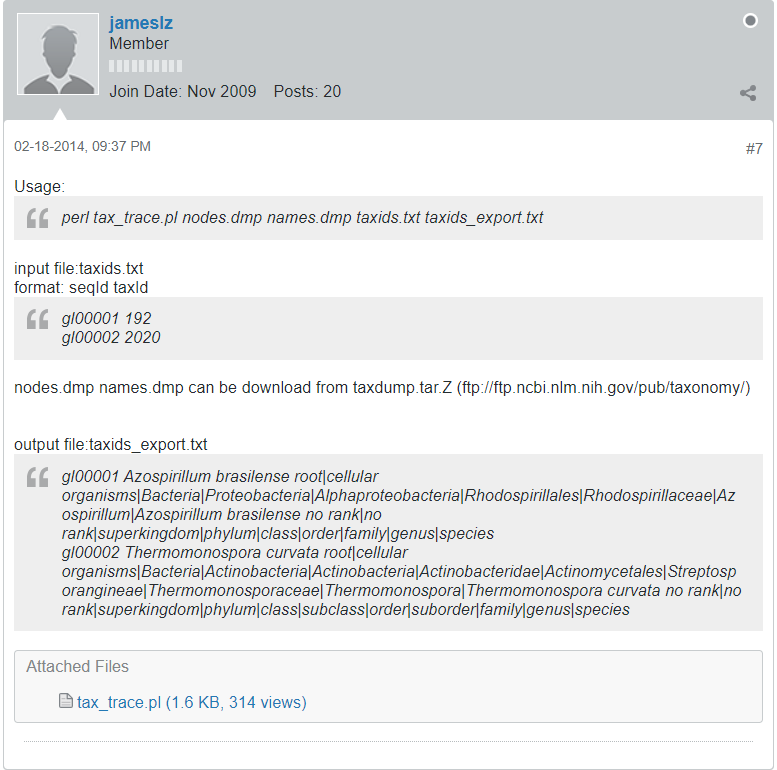
안타깝게도 이에 대한 해결책과 본인이 만든 perl 스크립트를 공유해 준 jameslz 님이 있지만, 스크립트 파일은 현재 다운 불가능이다.
나중에 perl좀 배워봐야겠다.
🟦 TaxonKit 소개
이를 극복하기 위하여 새로운 도구를 사용해 보았다. 바로 TaxonKit - NCBI Taxonomy Toolkit이다.
- 공식 홈페이지: https://bioinf.shenwei.me/taxonkit/
- 논문: Shen, W., & Ren, H. (2021). TaxonKit: A practical and efficient NCBI taxonomy toolkit. Journal of genetics and genomics = Yi chuan xue bao, 48(9), 844–850. https://doi.org/10.1016/j.jgg.2021.03.006 (2024.08.15 기준 인용 232회)
- Github: https://github.com/shenwei356/taxonkit
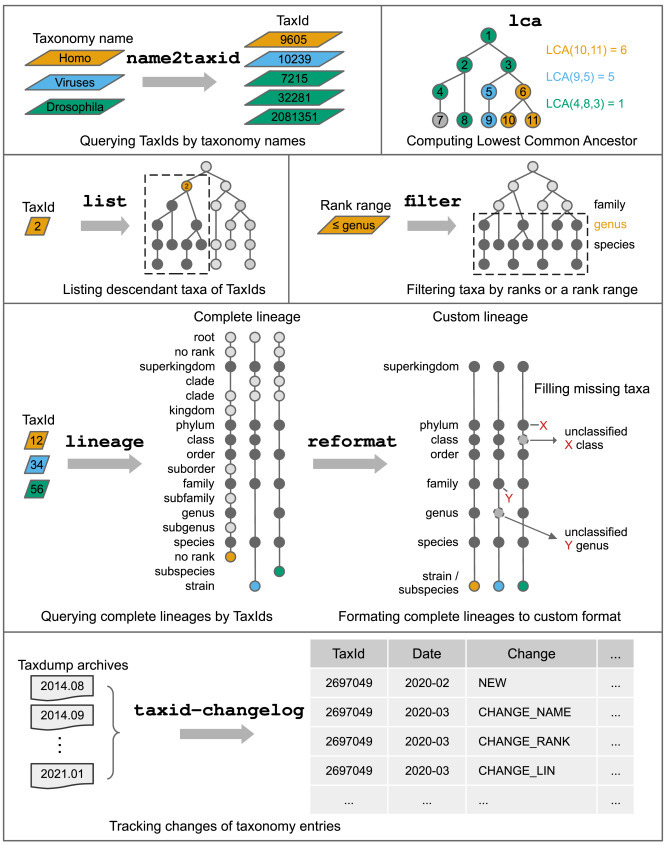
이 도구에서 제공하는 기능을 추리자면 아래와 같다.
1) taxonkit을 사용하여 tax id로 계통 정보를 추출한다
2) reformat으로 추출한 계통 정보를 subfamily, subgenus, stain 등 제거하고 깔끔하게 kingdom, phylum, class, order, family, Genus, Species만 추출 후 정리해 준다.
3) name2taxid를 사용하여 반대로 species이름으로 tax id를 추출할 수 있다.
🟦 설치
- https://bioinf.shenwei.me/taxonkit/download/
- 설치 방법은 1) 직접 다운로드 2) conda사용 3) home brew를 사용할 수 있다.
이 중에서 conda가 가장 최신 버전을 지원해 줌으로 conda로 설치해 보자.
conda install -c bioconda taxonkit
taxonkit -h # 설치 완료
그다음에는 계통 정보를 추출할 NCBI의 taxonomy database를 다운로드하여 보자.
cd ~/.taxonkit # 없다면 mkdir -p $HOME/.taxonkit 로 디렉토리 생성하기
wget ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz
tar -xvzf taxdump.tar.gz # 압축풀기
🟦 TaxonKit를 사용한 blast결과 정리
1) blast 돌리기
기존 blast 서열 output옵션에 tax id를 추가로 얻기 위하여 staxids를 추가해 보자,
blastn -db /data/Reference/BLASTDB/ITS_RefSeq_Fungi \
-query sequence.fasta \
-task blastn \
-dust no \
-outfmt "7 delim=, qacc staxids sacc evalue bitscore qcovus pident sscinames" \
-max_target_seqs 1 > blast-output
grep -v '#' blast-output > blast-output_summary
그렇다면 결과물의 두 번째 열에서 tax id를 찾을 수 있을 것이다.
2) tax_id 추출하기
두 번째 열에 존재하는 tax id를 추출하고 이를 저장해 보자.
awk -F',' '{print $2}'blast-output_summary > blast-output_taxid # tax_id만 추출
sed 's/.*;//' blast-output_taxid > blast-output_taxid2 # 간혹 tax_id가 ;로 두 숫자가 이어진 경우가 있음, 이때 두 번째 숫자만 추출
3) TaxonKit를 사용하여 전체 계통 정보 확인하기
taxonkit를 사용하여 lineage 정보를 추출하고, 이를 reformat을 사용하여 정리해 준다.
# tax_id에서 계통 정보 전체 추출하기
taxonkit lineage blast-output_taxid2 | awk '$2!=""' > blast_lineage.txt
# 추출된 정보 정리하기
cat blast_lineage.txt \
| taxonkit reformat -F \
| csvtk -H -t cut -f 1,3 \
| csvtk -H -t sep -f 2 -s ';' -R \
| csvtk add-header -t -n taxid,kindom,phylum,class,order,family,genus,species \
| csvtk pretty -ttaxonkit reformat -F에서 -F옵션은 빈칸을 "unclassified + 상위 tax level"으로 바꾸어 주는 기능입니다.
아래와 같은 결과물을 얻을 수 있습니다.

더 다양한 옵션을 알고 싶다면, 공식 튜토리얼을 참고하길 바랍니다!
=> https://bioinf.shenwei.me/taxonkit/usage/