

🟦 1. 서론
일단 데이터 분석의 자동화가 가능한가? 이는 데이터마다 다르다. 데이터 별로 각 EDA분석 이후 데이터의 품질을 보고 그 이후 분석 방법을 설계해야 한다. 그러나 마이크로바이옴 데이터의 경우 OTU table이라는 정형화된 데이터 형식이 있으며, 각 퀄리티가 떨어지는 데이터를 제외하고 분석하는 경우가 많아 이러한 변수의 영향을 덜 받는다고 말할 수 있다. 그러므로, 각 분석의 반복적 작업 단계를 자동화하는 것이 목표이며, 이에 대한 방법을 고민하고 있다.
Taxonomy 함수를 그릴 때 기본적인 R base의 색으로 표현해도 문제는 없지만, 외부 발표용 자료는 어느 정도 보는 사람이 잘 이해하도록 만들어야 한다. 하지만 수동적으로 색을 부과하는 작업은 시간이 낭비된다. 그래서 입력한 숫자에 따른 색 코드를 생산하는 Rcolorbrewer패키지를 사용해 보지만, 이도 기본적으로는 입력 단계를 필요로 한다.
시간소요를 줄이고자 taxonomy plot에 편리하게 색 코드를 뽑는 방법을 "Taxonomy composition plot에서 Phylum별로 Genus에 색 변화 주는 3가지 방법"에 나열하였다. 위 글에 나오는 "fantaxtic" 패키지를 이용하면 가장 빠르게 그림을 그릴 수 있다. 그러나 이는 색을 눈으로 구별하기 어렵다는 단점이 있었다.
또한 fantaxtic 패키지에서 Top 15의 taxa를 뽑아서 볼 때, 나타낸 Phylum에서 Top level을 제외한 Phylum은 모두 Others로 처리되는 단점이 있다.
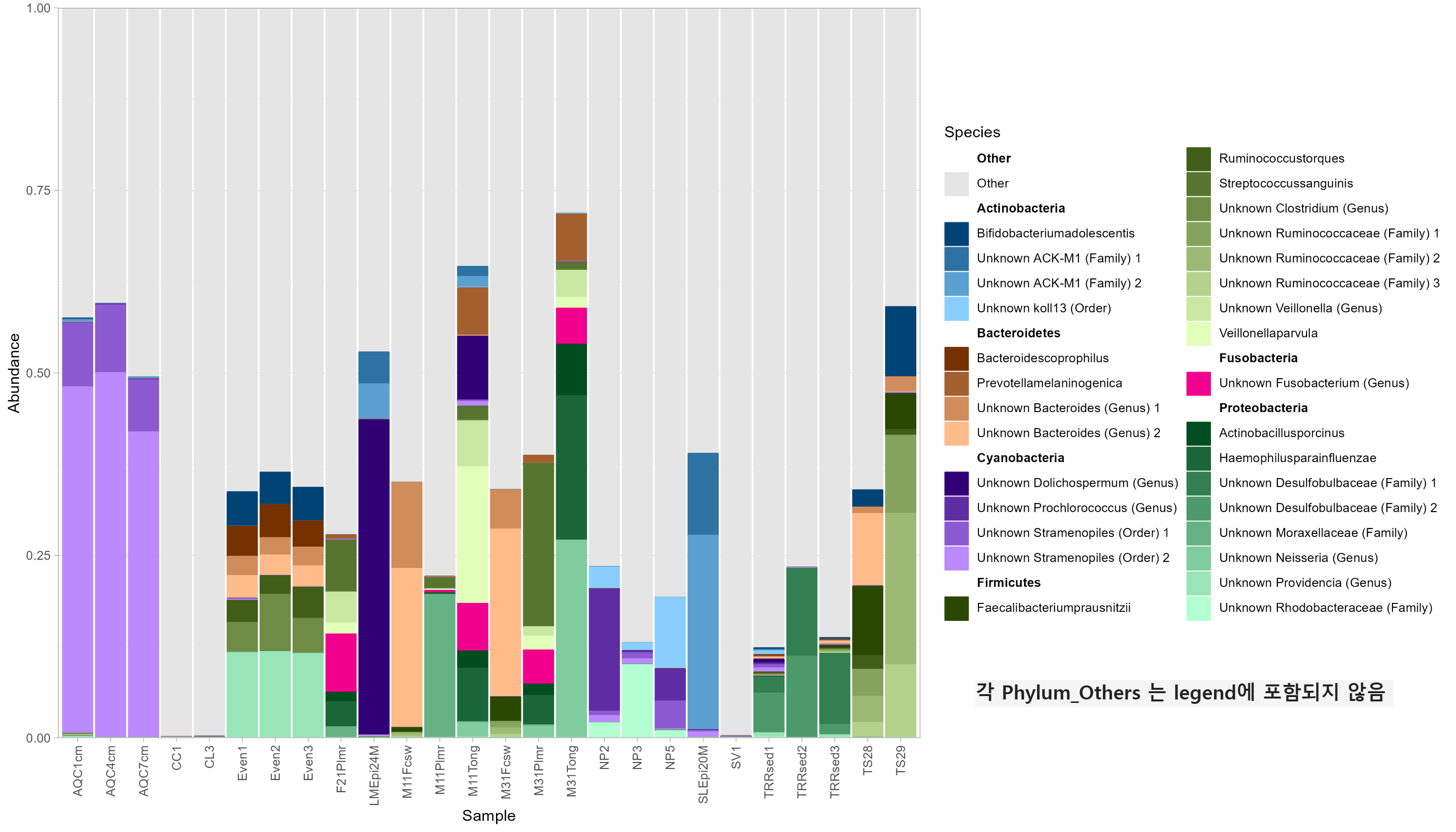
이러한 단점을 상쇄하기 위한 자동화 함수를 만들고자 하였고, 여러 조건들을 생각해 보았다.
1. Top N 수에 따라 표현
보고 싶은 taxa의 Top N 수를 입력하면, 이에 따른 Phylum과, 각 Phylum에서 Top N에 포함되지 않는 taxa는 모두 Phylum_Other처리하여 표현한다.
2. 각 Phylum별로 색 지정
피부 마이크로바이옴의 경우 Phylum 수준에서 Actinobacteria와 Firmicutes에 속하는 미생물에 관한 연구가 많다. 이를 위해 각 색을 대비되는 빨강과 파랑으로 지정하고, 그 다음으로 많은 비율을 차지하는 Bacteroidetes, Proteobacteria, Fusobacteria의 색도 보라, 초록, 주황색으로 지정하였다. 각 색의 분주는 Rcolorbrewer 패키지의 색을 사용하고, 나머지 phylum는 랜덤으로 5개 정도 추가하였다. Top n 수를 보여주는 경우 많은 종류의 Phylum이 나오지 않음으로, 5개 정도 색의 추가로 충분할 것이라 생각하였다.
하지만 Rcolorbrewer 패키지의 경우, 연속된 색 팔레트는 9종류 이상 표현하지 않는 것이 많다. 이를 고려하여 9 색 이상의 경우는 R base의 heat.colors()를 사용하거나 직접 색을 입력하는 colorRampPalette()등을 활용하였다.
3. Top level은 Phylum이며, 그다음 보여줄 level은 지정
그림에 표현되는 Top taxa는 Phylum이지만, 각 색을 지정할 taxa는 선택 가능 하도록 하였다.
위 내용을 기준으로 완성한 함수는 아래와 같다.
🟦 2. 자동화 함수 : tax_barplot_RBrewer
[input]
.melt. = phyloseq에서 psmelt를 이용해 분해된 데이터
.taxa. = 보여주고자 하는 taxa level
.top. = 보고자 하는 taxa level에서 상위 n 수를 나타내겠다.
[output]
output = 리스트 형태의 데이터
output$Finalcolor = 그림에서 사용할 색 코드
output$data = 변환된 데이터
4단계로 이루어져 있으며 각 단계는 아래와 같다.
1. Top N taxa추출하기
2. Top N을 제외하고 Other로 변경하기
3. 샘플 정렬하기
- Phylum은 ABC순으로 정렬하였으며 Phylum내의 taxa는 상대 풍부도가 높은 순으로 정렬
4. 각 Phylum마다 색 분주하기
5. output 데이터 변환하기
tax_barplot_RBrewer <- function(.melt., .taxa., .top.) {
# 1. select Top taxa ____________________________________________________________
Order = tapply(.melt.$Abundance, .melt.[, .taxa.], sum) %>% sort(decreasing = T)
Top_g <- names(Order[c(1:.top.)]) # TOP N에 해당하는 Genus
Top_p <- .melt.[.melt.[, .taxa.] %in% Top_g, "Phylum"]%>% unique() # TOP N에 해당하는 Genus의 Phylum
p_tax_table <- .melt.[.melt.[, .taxa.] %in% Top_g, c("Phylum", .taxa.)] %>%
.[!duplicated(.[ , .taxa.]),] # Top N에 해당하는 taxa를 테이블로 정리
# 2. define Others _____________________________________________________________
.melt.2 <- .melt. # Back up
# Not Topn taxa <- Others
.melt.2[!.melt.2[, "Phylum"] %in% Top_p, "Phylum"] <- "Others"
.melt.2[!.melt.2[, "Phylum"] %in% Top_p, .taxa.] <- "Others"
# Not Top_n taxa -> Others + Phylum
for ( i in Top_p ) {
G <- p_tax_table[p_tax_table[, "Phylum" ] == i , .taxa.]
.melt.2[.melt.2[, "Phylum" ] == i &
!.melt.2[, .taxa.] %in% G, .taxa. ] <- paste0(i, "_Others")
}
# Top_n taxa -> Phylum_taxa name format
for ( i in Top_p ) {
G <- p_tax_table[p_tax_table[, "Phylum"] == i , .taxa.]
for (g in G){
.melt.2[.melt.2[, .taxa.] == g, .taxa.] <- paste0(i,"_", g)
}
}
# check
.melt.2[.melt.2[, "Phylum"] %in% Top_p ,.taxa.] %>% unique()
# 3. order _____________________________________________________________________
.melt.3 <- .melt.2 # 백업
# Genus order by phylum and Abundance
table <- .melt.3[.melt.3[, "Phylum" ] %in% Top_p, c("Abundance", "Phylum", .taxa.)]
table.2 <- table %>% group_by_(.dots = "Phylum", .taxa.) %>%
dplyr::summarise(sum.Abundance=sum(Abundance), .groups = 'drop') %>%
as.data.frame()
g_order <- table.2 %>% arrange( desc(sum.Abundance)) %>% arrange_("Phylum") %>% .[,.taxa. ] # No
p_order <- table %>% .[,"Phylum" ]%>% unique %>% sort
# ordering Genus column
.melt.4 <- .melt.3
.melt.4[,"Phylum"] <- factor(.melt.4[,"Phylum" ], levels = p_order)
.melt.4[,.taxa.] <- factor(.melt.4[,.taxa.], levels = c(g_order, "Others"))
# check
.melt.4[,.taxa.]%>% levels()
# 4. palette ___________________________________________________________________
table.3 <-table.2 %>% arrange( desc(sum.Abundance)) %>% arrange_("Phylum") %>% select_("Phylum", .taxa.) # No
df <- table.3
# Find how many colour categories to create and the number of colours in each
categories <- aggregate(as.formula(paste(.taxa., "Phylum", sep="~" )), df,
function(x) length(unique(x)))
# Create a color-coded print list
color_list.names <- categories[, "Phylum"]
color_list <- vector("list", length(color_list.names))
names(color_list) <- color_list.names
# https://stackoverflow.com/questions/5688020/how-to-create-a-list-with-names-but-no-entries-in-r-splus
'%!in%' <- function(x,y)!('%in%'(x,y))
P <- categories$Phylum
basic_p <-c("Bacteroidetes", "Proteobacteria", "Firmicutes", "Fusobacteria", "Actinobacteria")
# for 문
for (i in P) {
if (any(i == "Actinobacteria")) {
Ac_num <- categories[categories[, "Phylum"] == "Actinobacteria", .taxa.]
ifelse(Ac_num == 1, Actinobacteria_color <- rev(brewer.pal(9, "Reds")[5]),
ifelse(Ac_num == 2, Actinobacteria_color <- rev(brewer.pal(9, "Reds")[c(2, 7)]),
ifelse(Ac_num >=3 & Ac_num <= 9, Actinobacteria_color <- rev(brewer.pal(Ac_num, "Reds")),
Actinobacteria_color <- heat.colors(Ac_num)))) # heat.colors : Red ~ yellow
color_list$Actinobacteria <- Actinobacteria_color
} else if (i == "Bacteroidetes") {
Ba_num <- categories[categories[, "Phylum"] == "Bacteroidetes", .taxa.]
ifelse(Ba_num == 1, Bacteroidetes_color <- rev(brewer.pal(9, "Purples")[5]),
ifelse(Ba_num == 2, Bacteroidetes_color <- rev(brewer.pal(9, "Purples")[c(3, 7)]),
ifelse(Ba_num >=3 & Ba_num <= 9, Bacteroidetes_color <- rev(brewer.pal(Ba_num, "Purples")),
Bacteroidetes_color <- cm.colors(Ba_num)))) # cm.colors : Pink ~ Blue
color_list$Bacteroidetes <- Bacteroidetes_color
} else if (i == "Proteobacteria") {
Pr_num <- categories[categories[, "Phylum"] == "Proteobacteria", .taxa.]
ifelse(Pr_num == 1, Proteobacteria_color <- rev(brewer.pal(9, "Greens")[5]),
ifelse(Pr_num == 2, Proteobacteria_color <- rev(brewer.pal(9, "Greens")[c(3, 7)]),
ifelse(Pr_num >=3 & Pr_num <= 9, Proteobacteria_color <- rev(brewer.pal(Pr_num, "Greens")),
Proteobacteria_color <- topo.colors(Pr_num)))) # topo.colors : yellow ~ Blue
color_list$Proteobacteria <- Proteobacteria_color
} else if (i == "Firmicutes") {
Fi_num <- categories[categories[, "Phylum"] == "Firmicutes", .taxa.]
ifelse(Fi_num == 1, Firmicutes_color <- rev(brewer.pal(9, "Blues")[5]),
ifelse(Fi_num == 2, Firmicutes_color <- rev(brewer.pal(9, "Blues")[c(3, 7)]),
ifelse(Fi_num >=3 & Fi_num <= 9, Firmicutes_color <- rev(brewer.pal(Fi_num, "Blues")),
Firmicutes_color <- terrain.colors(Fi_num)))) # cm.colors : Pink ~ Blue
color_list$Firmicutes <- Firmicutes_color
} else if (i == "Fusobacteria") {
Fu_num <- categories[categories[, "Phylum"] == "Fusobacteria", .taxa.]
B_to_O <- colorRampPalette(c("skyblue", "orange"))
ifelse(Fu_num == 1, Fusobacteria_color <- rev(brewer.pal(9, "YlOrBr")[5]),
ifelse(Fu_num == 2, Fusobacteria_color <- rev(brewer.pal(9, "YlOrBr")[c(3, 7)]),
ifelse(Fu_num >=3 & Fu_num <= 9, Fusobacteria_color <- rev(brewer.pal(Fu_num, "YlOrBr")),
Fusobacteria_color <- B_to_O(Fu_num)))) # cm.colors : Pink ~ Blue
color_list$Fusobacteria <- Fusobacteria_color
} else if (i %!in% basic_p) {
other_p <- categories[categories$Phylum %!in% c("Bacteroidetes", "Proteobacteria", "Firmicutes",
"Fusobacteria", "Actinobacteria"), "Phylum"]
others_colors <- c("RdPu", "YlOrBr", "Spectral", "BrBG", "PuOr")
for ( i in 1:length(other_p) ) {
p <- other_p[i]
color <- others_colors[i]
num <- categories[categories$Phylum == p, .taxa.]
ifelse(num == 1, col <- rev(brewer.pal(9, color)[5]),
ifelse(num == 2, col <- rev(brewer.pal(9, color)[c(3, 7)]),
ifelse(num >=3 & num <= 9, col <- rev(brewer.pal(num, color)),
stop("Number of Other Phylum is more than 9, Use manual way.")
)))
name <- paste0(p, "_color")
assign(name, col)
color_list[[p]] <- get(name)
}
}
}
# 5. output ___________________________________________________________________
color_vector <- color_list %>% unlist %>% unname
Final_col <- c(color_vector, "#D3D3D3") # Other에 해당하는 회색 추가
return(list(Final_color=Final_col, data=.melt.4))
}
🟦 3. 함수 적용
위 함수를 이용해 taxonomy plot을 그려보자
예제 데이터는 Qiime2의 moving-pictures데이터이다.
# 패키지 불러오기
library(tidyverse)
library(phyloseq)
library(RColorBrewer)
# 데이터 불러오기
ps <- readRDS("./ps.rds")
ps.rel <- transform_sample_counts(ps, function(x) x / sum(x) )
melt <- ps.rel %>% tax_glom(taxrank = "Genus") %>% psmelt()
# 함수 돌리기
output <- tax_barplot_RBrewer(.melt. = melt,
.taxa. = "Genus",
.top. = 20)
output$Final_color
output$data
# 샘플 순서 지정하기
sample_order <- output$data %>%
data.frame() %>%
# Calculate relative abundances
dplyr::group_by(Sample) %>%
mutate(Abundance = Abundance / sum(Abundance)) %>%
# Sort by taxon of interest
filter(Phylum == "Proteobacteria") %>%
dplyr::group_by(Sample) %>%
dplyr::summarise(Abundance = sum(Abundance)) %>%
dplyr::arrange(Abundance) %>%
# Extract the sample order
pull(Sample) %>%
as.character()
df <- output$data
df$Sample <- factor(df$Sample, levels = sample_order)
# Plot 그리기
p <- ggplot(data=df, aes(x=Sample, y=Abundance, fill=Genus)) +
geom_bar(aes(), position="fill", stat="identity", colour=NA ) +
theme_classic() +
theme(legend.position="bottom" ,
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_blank()) +
labs(y = "Relative Abundance (%)") +
theme(axis.text.x.bottom = element_text(angle = 45, vjust = 1, hjust=1),
axis.text = element_text(size = 7),
legend.position = "right")
p
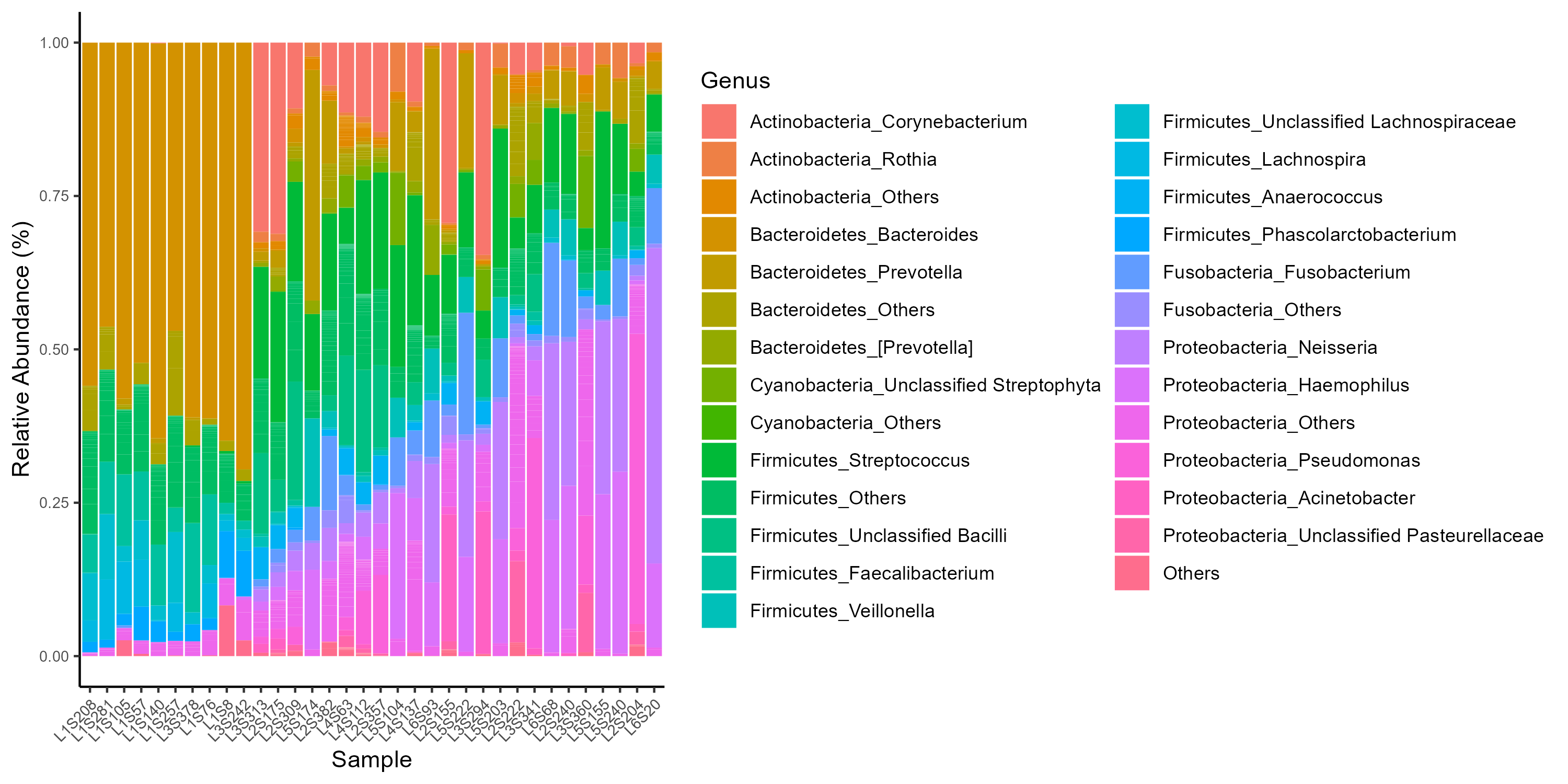
일단 함수에서 만들어진 색 코드를 적용하기 전이다. R에서 기본적으로 적용되는 무지개 색이 적용되었다.
각 Phlyum에서 Top 20에 해당하는 Genus와 각 Phylum의 Other도 표현되었다. 사실 Other를 Others로 잘못 적긴 했지만 이 글을 적는 지금에서야 알아차렸다.
"ggnested" 패키지의 함수처럼 각 Legend를 Phylum 별로 나누는 것도 좋지만, 단계가 복잡하여 따로 추가하지 않았다.

p + scale_fill_manual(name ='Phylum - Genus', values = output$Final_color)
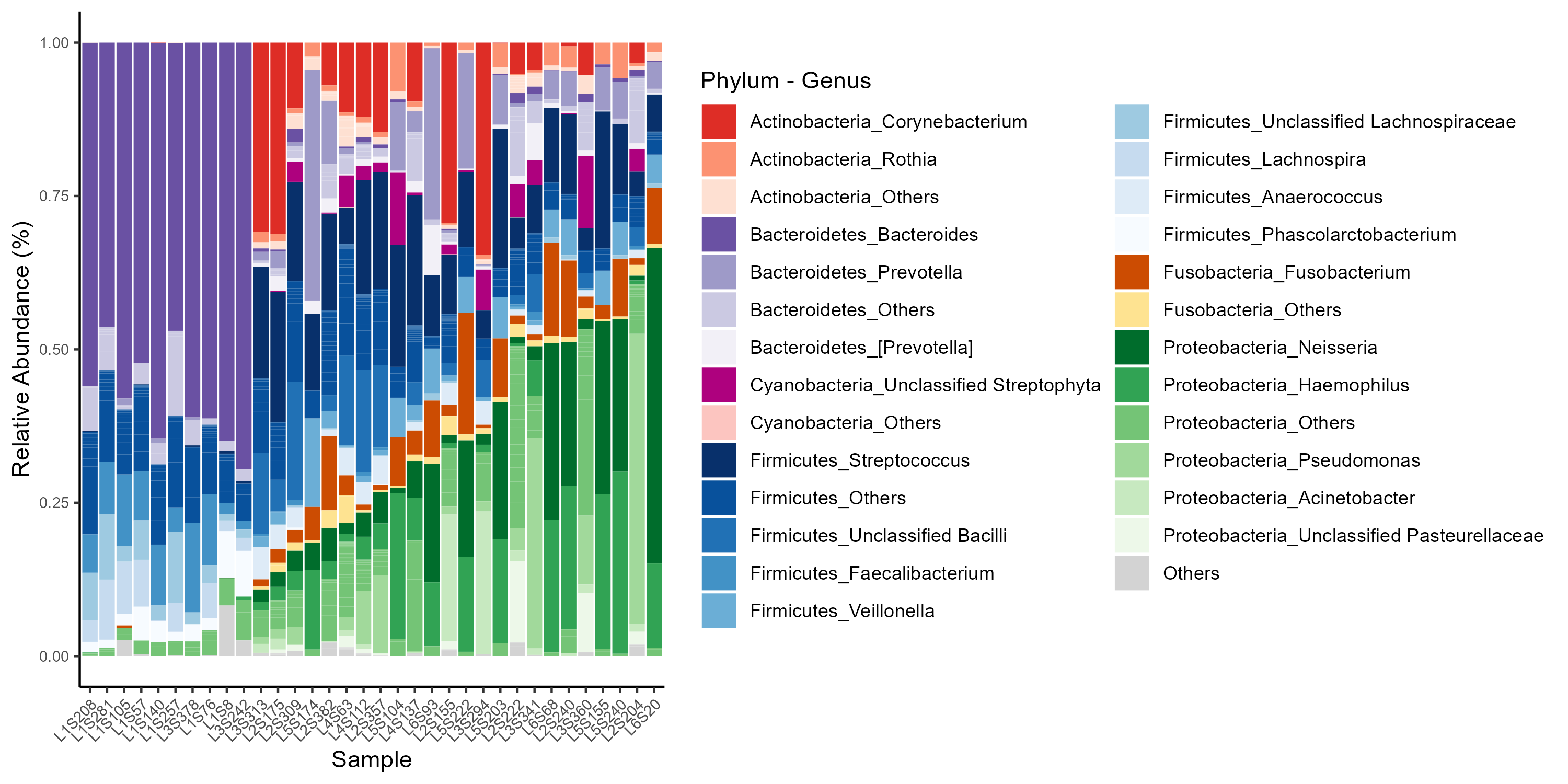
output$Final_color를 적용한 모습이다.
각 Phylum에 맞게 색이 지정되었으며, 각 Phylum에 Other도 잘 표현되었음을 알 수 있다.
또한 plot을 그리기 전에 proteobacteria의 풍부도 순으로 정렬된 것도 한눈에 잘 볼 수 있다.
하지만 이상하게 실제 데이터에 적용하려고 하면 오류가 난다...
다시 한번 점검을 해봐야 겠다.

🟦 1. 서론
일단 데이터 분석의 자동화가 가능한가? 이는 데이터마다 다르다. 데이터 별로 각 EDA분석 이후 데이터의 품질을 보고 그 이후 분석 방법을 설계해야 한다. 그러나 마이크로바이옴 데이터의 경우 OTU table이라는 정형화된 데이터 형식이 있으며, 각 퀄리티가 떨어지는 데이터를 제외하고 분석하는 경우가 많아 이러한 변수의 영향을 덜 받는다고 말할 수 있다. 그러므로, 각 분석의 반복적 작업 단계를 자동화하는 것이 목표이며, 이에 대한 방법을 고민하고 있다.
Taxonomy 함수를 그릴 때 기본적인 R base의 색으로 표현해도 문제는 없지만, 외부 발표용 자료는 어느 정도 보는 사람이 잘 이해하도록 만들어야 한다. 하지만 수동적으로 색을 부과하는 작업은 시간이 낭비된다. 그래서 입력한 숫자에 따른 색 코드를 생산하는 Rcolorbrewer패키지를 사용해 보지만, 이도 기본적으로는 입력 단계를 필요로 한다.
시간소요를 줄이고자 taxonomy plot에 편리하게 색 코드를 뽑는 방법을 "Taxonomy composition plot에서 Phylum별로 Genus에 색 변화 주는 3가지 방법"에 나열하였다. 위 글에 나오는 "fantaxtic" 패키지를 이용하면 가장 빠르게 그림을 그릴 수 있다. 그러나 이는 색을 눈으로 구별하기 어렵다는 단점이 있었다.
또한 fantaxtic 패키지에서 Top 15의 taxa를 뽑아서 볼 때, 나타낸 Phylum에서 Top level을 제외한 Phylum은 모두 Others로 처리되는 단점이 있다.
이러한 단점을 상쇄하기 위한 자동화 함수를 만들고자 하였고, 여러 조건들을 생각해 보았다.
1. Top N 수에 따라 표현
보고 싶은 taxa의 Top N 수를 입력하면, 이에 따른 Phylum과, 각 Phylum에서 Top N에 포함되지 않는 taxa는 모두 Phylum_Other처리하여 표현한다.
2. 각 Phylum별로 색 지정
피부 마이크로바이옴의 경우 Phylum 수준에서 Actinobacteria와 Firmicutes에 속하는 미생물에 관한 연구가 많다. 이를 위해 각 색을 대비되는 빨강과 파랑으로 지정하고, 그 다음으로 많은 비율을 차지하는 Bacteroidetes, Proteobacteria, Fusobacteria의 색도 보라, 초록, 주황색으로 지정하였다. 각 색의 분주는 Rcolorbrewer 패키지의 색을 사용하고, 나머지 phylum는 랜덤으로 5개 정도 추가하였다. Top n 수를 보여주는 경우 많은 종류의 Phylum이 나오지 않음으로, 5개 정도 색의 추가로 충분할 것이라 생각하였다.
하지만 Rcolorbrewer 패키지의 경우, 연속된 색 팔레트는 9종류 이상 표현하지 않는 것이 많다. 이를 고려하여 9 색 이상의 경우는 R base의 heat.colors()를 사용하거나 직접 색을 입력하는 colorRampPalette()등을 활용하였다.
3. Top level은 Phylum이며, 그다음 보여줄 level은 지정
그림에 표현되는 Top taxa는 Phylum이지만, 각 색을 지정할 taxa는 선택 가능 하도록 하였다.
위 내용을 기준으로 완성한 함수는 아래와 같다.
🟦 2. 자동화 함수 : tax_barplot_RBrewer
[input]
.melt. = phyloseq에서 psmelt를 이용해 분해된 데이터
.taxa. = 보여주고자 하는 taxa level
.top. = 보고자 하는 taxa level에서 상위 n 수를 나타내겠다.
[output]
output = 리스트 형태의 데이터
output$Finalcolor = 그림에서 사용할 색 코드
output$data = 변환된 데이터
4단계로 이루어져 있으며 각 단계는 아래와 같다.
1. Top N taxa추출하기
2. Top N을 제외하고 Other로 변경하기
3. 샘플 정렬하기
- Phylum은 ABC순으로 정렬하였으며 Phylum내의 taxa는 상대 풍부도가 높은 순으로 정렬
4. 각 Phylum마다 색 분주하기
5. output 데이터 변환하기
tax_barplot_RBrewer <- function(.melt., .taxa., .top.) {
# 1. select Top taxa ____________________________________________________________
Order = tapply(.melt.$Abundance, .melt.[, .taxa.], sum) %>% sort(decreasing = T)
Top_g <- names(Order[c(1:.top.)]) # TOP N에 해당하는 Genus
Top_p <- .melt.[.melt.[, .taxa.] %in% Top_g, "Phylum"]%>% unique() # TOP N에 해당하는 Genus의 Phylum
p_tax_table <- .melt.[.melt.[, .taxa.] %in% Top_g, c("Phylum", .taxa.)] %>%
.[!duplicated(.[ , .taxa.]),] # Top N에 해당하는 taxa를 테이블로 정리
# 2. define Others _____________________________________________________________
.melt.2 <- .melt. # Back up
# Not Topn taxa <- Others
.melt.2[!.melt.2[, "Phylum"] %in% Top_p, "Phylum"] <- "Others"
.melt.2[!.melt.2[, "Phylum"] %in% Top_p, .taxa.] <- "Others"
# Not Top_n taxa -> Others + Phylum
for ( i in Top_p ) {
G <- p_tax_table[p_tax_table[, "Phylum" ] == i , .taxa.]
.melt.2[.melt.2[, "Phylum" ] == i &
!.melt.2[, .taxa.] %in% G, .taxa. ] <- paste0(i, "_Others")
}
# Top_n taxa -> Phylum_taxa name format
for ( i in Top_p ) {
G <- p_tax_table[p_tax_table[, "Phylum"] == i , .taxa.]
for (g in G){
.melt.2[.melt.2[, .taxa.] == g, .taxa.] <- paste0(i,"_", g)
}
}
# check
.melt.2[.melt.2[, "Phylum"] %in% Top_p ,.taxa.] %>% unique()
# 3. order _____________________________________________________________________
.melt.3 <- .melt.2 # 백업
# Genus order by phylum and Abundance
table <- .melt.3[.melt.3[, "Phylum" ] %in% Top_p, c("Abundance", "Phylum", .taxa.)]
table.2 <- table %>% group_by_(.dots = "Phylum", .taxa.) %>%
dplyr::summarise(sum.Abundance=sum(Abundance), .groups = 'drop') %>%
as.data.frame()
g_order <- table.2 %>% arrange( desc(sum.Abundance)) %>% arrange_("Phylum") %>% .[,.taxa. ] # No
p_order <- table %>% .[,"Phylum" ]%>% unique %>% sort
# ordering Genus column
.melt.4 <- .melt.3
.melt.4[,"Phylum"] <- factor(.melt.4[,"Phylum" ], levels = p_order)
.melt.4[,.taxa.] <- factor(.melt.4[,.taxa.], levels = c(g_order, "Others"))
# check
.melt.4[,.taxa.]%>% levels()
# 4. palette ___________________________________________________________________
table.3 <-table.2 %>% arrange( desc(sum.Abundance)) %>% arrange_("Phylum") %>% select_("Phylum", .taxa.) # No
df <- table.3
# Find how many colour categories to create and the number of colours in each
categories <- aggregate(as.formula(paste(.taxa., "Phylum", sep="~" )), df,
function(x) length(unique(x)))
# Create a color-coded print list
color_list.names <- categories[, "Phylum"]
color_list <- vector("list", length(color_list.names))
names(color_list) <- color_list.names
# https://stackoverflow.com/questions/5688020/how-to-create-a-list-with-names-but-no-entries-in-r-splus
'%!in%' <- function(x,y)!('%in%'(x,y))
P <- categories$Phylum
basic_p <-c("Bacteroidetes", "Proteobacteria", "Firmicutes", "Fusobacteria", "Actinobacteria")
# for 문
for (i in P) {
if (any(i == "Actinobacteria")) {
Ac_num <- categories[categories[, "Phylum"] == "Actinobacteria", .taxa.]
ifelse(Ac_num == 1, Actinobacteria_color <- rev(brewer.pal(9, "Reds")[5]),
ifelse(Ac_num == 2, Actinobacteria_color <- rev(brewer.pal(9, "Reds")[c(2, 7)]),
ifelse(Ac_num >=3 & Ac_num <= 9, Actinobacteria_color <- rev(brewer.pal(Ac_num, "Reds")),
Actinobacteria_color <- heat.colors(Ac_num)))) # heat.colors : Red ~ yellow
color_list$Actinobacteria <- Actinobacteria_color
} else if (i == "Bacteroidetes") {
Ba_num <- categories[categories[, "Phylum"] == "Bacteroidetes", .taxa.]
ifelse(Ba_num == 1, Bacteroidetes_color <- rev(brewer.pal(9, "Purples")[5]),
ifelse(Ba_num == 2, Bacteroidetes_color <- rev(brewer.pal(9, "Purples")[c(3, 7)]),
ifelse(Ba_num >=3 & Ba_num <= 9, Bacteroidetes_color <- rev(brewer.pal(Ba_num, "Purples")),
Bacteroidetes_color <- cm.colors(Ba_num)))) # cm.colors : Pink ~ Blue
color_list$Bacteroidetes <- Bacteroidetes_color
} else if (i == "Proteobacteria") {
Pr_num <- categories[categories[, "Phylum"] == "Proteobacteria", .taxa.]
ifelse(Pr_num == 1, Proteobacteria_color <- rev(brewer.pal(9, "Greens")[5]),
ifelse(Pr_num == 2, Proteobacteria_color <- rev(brewer.pal(9, "Greens")[c(3, 7)]),
ifelse(Pr_num >=3 & Pr_num <= 9, Proteobacteria_color <- rev(brewer.pal(Pr_num, "Greens")),
Proteobacteria_color <- topo.colors(Pr_num)))) # topo.colors : yellow ~ Blue
color_list$Proteobacteria <- Proteobacteria_color
} else if (i == "Firmicutes") {
Fi_num <- categories[categories[, "Phylum"] == "Firmicutes", .taxa.]
ifelse(Fi_num == 1, Firmicutes_color <- rev(brewer.pal(9, "Blues")[5]),
ifelse(Fi_num == 2, Firmicutes_color <- rev(brewer.pal(9, "Blues")[c(3, 7)]),
ifelse(Fi_num >=3 & Fi_num <= 9, Firmicutes_color <- rev(brewer.pal(Fi_num, "Blues")),
Firmicutes_color <- terrain.colors(Fi_num)))) # cm.colors : Pink ~ Blue
color_list$Firmicutes <- Firmicutes_color
} else if (i == "Fusobacteria") {
Fu_num <- categories[categories[, "Phylum"] == "Fusobacteria", .taxa.]
B_to_O <- colorRampPalette(c("skyblue", "orange"))
ifelse(Fu_num == 1, Fusobacteria_color <- rev(brewer.pal(9, "YlOrBr")[5]),
ifelse(Fu_num == 2, Fusobacteria_color <- rev(brewer.pal(9, "YlOrBr")[c(3, 7)]),
ifelse(Fu_num >=3 & Fu_num <= 9, Fusobacteria_color <- rev(brewer.pal(Fu_num, "YlOrBr")),
Fusobacteria_color <- B_to_O(Fu_num)))) # cm.colors : Pink ~ Blue
color_list$Fusobacteria <- Fusobacteria_color
} else if (i %!in% basic_p) {
other_p <- categories[categories$Phylum %!in% c("Bacteroidetes", "Proteobacteria", "Firmicutes",
"Fusobacteria", "Actinobacteria"), "Phylum"]
others_colors <- c("RdPu", "YlOrBr", "Spectral", "BrBG", "PuOr")
for ( i in 1:length(other_p) ) {
p <- other_p[i]
color <- others_colors[i]
num <- categories[categories$Phylum == p, .taxa.]
ifelse(num == 1, col <- rev(brewer.pal(9, color)[5]),
ifelse(num == 2, col <- rev(brewer.pal(9, color)[c(3, 7)]),
ifelse(num >=3 & num <= 9, col <- rev(brewer.pal(num, color)),
stop("Number of Other Phylum is more than 9, Use manual way.")
)))
name <- paste0(p, "_color")
assign(name, col)
color_list[[p]] <- get(name)
}
}
}
# 5. output ___________________________________________________________________
color_vector <- color_list %>% unlist %>% unname
Final_col <- c(color_vector, "#D3D3D3") # Other에 해당하는 회색 추가
return(list(Final_color=Final_col, data=.melt.4))
}
🟦 3. 함수 적용
위 함수를 이용해 taxonomy plot을 그려보자
예제 데이터는 Qiime2의 moving-pictures데이터이다.
# 패키지 불러오기
library(tidyverse)
library(phyloseq)
library(RColorBrewer)
# 데이터 불러오기
ps <- readRDS("./ps.rds")
ps.rel <- transform_sample_counts(ps, function(x) x / sum(x) )
melt <- ps.rel %>% tax_glom(taxrank = "Genus") %>% psmelt()
# 함수 돌리기
output <- tax_barplot_RBrewer(.melt. = melt,
.taxa. = "Genus",
.top. = 20)
output$Final_color
output$data
# 샘플 순서 지정하기
sample_order <- output$data %>%
data.frame() %>%
# Calculate relative abundances
dplyr::group_by(Sample) %>%
mutate(Abundance = Abundance / sum(Abundance)) %>%
# Sort by taxon of interest
filter(Phylum == "Proteobacteria") %>%
dplyr::group_by(Sample) %>%
dplyr::summarise(Abundance = sum(Abundance)) %>%
dplyr::arrange(Abundance) %>%
# Extract the sample order
pull(Sample) %>%
as.character()
df <- output$data
df$Sample <- factor(df$Sample, levels = sample_order)
# Plot 그리기
p <- ggplot(data=df, aes(x=Sample, y=Abundance, fill=Genus)) +
geom_bar(aes(), position="fill", stat="identity", colour=NA ) +
theme_classic() +
theme(legend.position="bottom" ,
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_blank()) +
labs(y = "Relative Abundance (%)") +
theme(axis.text.x.bottom = element_text(angle = 45, vjust = 1, hjust=1),
axis.text = element_text(size = 7),
legend.position = "right")
p
일단 함수에서 만들어진 색 코드를 적용하기 전이다. R에서 기본적으로 적용되는 무지개 색이 적용되었다.
각 Phlyum에서 Top 20에 해당하는 Genus와 각 Phylum의 Other도 표현되었다. 사실 Other를 Others로 잘못 적긴 했지만 이 글을 적는 지금에서야 알아차렸다.
"ggnested" 패키지의 함수처럼 각 Legend를 Phylum 별로 나누는 것도 좋지만, 단계가 복잡하여 따로 추가하지 않았다.
p + scale_fill_manual(name ='Phylum - Genus', values = output$Final_color)
output$Final_color를 적용한 모습이다.
각 Phylum에 맞게 색이 지정되었으며, 각 Phylum에 Other도 잘 표현되었음을 알 수 있다.
또한 plot을 그리기 전에 proteobacteria의 풍부도 순으로 정렬된 것도 한눈에 잘 볼 수 있다.
하지만 이상하게 실제 데이터에 적용하려고 하면 오류가 난다...
다시 한번 점검을 해봐야 겠다.