
작성 : 2023.04.13~2023-04-17
수정: 2024-02-05
picrust2 visualization

들어가기에 앞서
오늘은 올해 따끈따끈하게 출시된(무려 2023년 4월 8일) ggpicrust2 패키지를 소개합니다. ggpicrust2는 마이크바이옴의 기능 예측 도구인 picrust2의 결과물을 통계적으로 분석하고 시각화하는 데에 사용합니다.
> 분석 환경
- biom과 picrust 설치 필요.
- 추가적으로 분석은 R환경 (최신 버전)
> 예제 데이터: QIIME2 예제인 moving-picture
- 이는 사람의 혀, 장, 양 손바닥의 마이크로바이옴 데이터를 담고 있다. 이 중에서 혀와 장의 마이크로바이옴에 해당하는 기능예측 유전자를 비교해 본다.
ggpicrust2
- 관련 논문 : https://arxiv.org/abs/2303.10388
- 깃허브 : https://github.com/cafferychen777/ggpicrust2 (사용 시 오류 있다면 깃허브의 issue에 문의)
- 공식 튜토리얼 : https://rpubs.com/cafferychen7850/1023044
1. PICRUSt2 시행하기
1) input 파일 준비
picrust2는 리눅스 상에서 분석 가능합니다. 사용가능한 예제 파일은 아래에 있으며, picrust2 결과 파일도 아래에 첨부해 두었으니, picrust2가 실행되지 않더라도 위 튜토리얼은 진행 가능합니다.
(1) FASTA 파일
(2) OTU table(feature table, ASV table)
otu table을 먼저 picrust에 사용되는 biom형식으로 변환해주어야 합니다 ( 관련 방법은 링크를 참고).
biom convert -i otu_for_picrust.txt -o otu_for_picrust.biom --table-type="OTU table" --to-json
2) PICRUSt2 돌리기
이제 변환된 biom파일과 fasta 파일을 이용해 picrust2를 실행합니다(picrust2의 설치는 링크를 참고).
conda activate picrust2
picrust2_pipeline.py -s sequences.fasta -i otu_for_picrust.biom -o picrust_result/분석이 완료되면 picrust_result/에 아래와 같은 파일들이 생성됩니다.
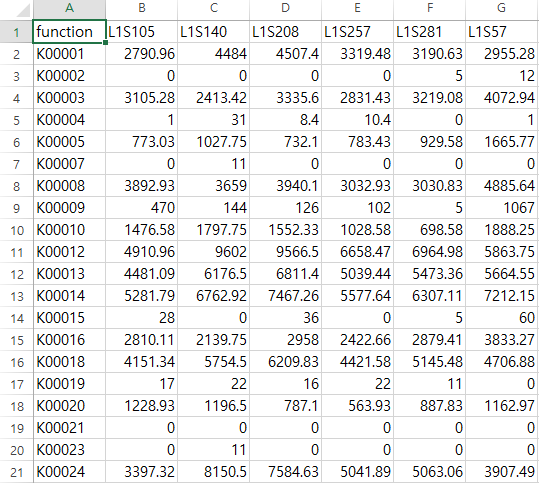
우리는 이 중에서 KO_metagenome_out폴더 안에 있는 pred_metagenome_unstrat.tsv.gz파일을 사용합니다. pred_metagenome_unstrat.tsv.gz파일의 압축을 풀어서 준비합니다.
파일을 열어보면 열 이름은 각 샘플의 이름이 적혀있고, 행의 이름에는 각 ko번호에 해당하는 id가 있습니다. 이는 otu table과 비슷한 형태입니다.
2. ggpicrust를 이용한 시각화
1) 패키지와 분석 데이터 불러오기
기본적인 패키지들이 설치되어있어야 합니다.
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c(
"ALDEx2", "DESeq2", "edgeR", "limma", "Maaslin2", "metagenomeSeq",
"SummarizedExperiment", "phyloseq", "biomformat", "lefser"
))
install.packages("ggpicrust2")
install.packages("ggprism")
install.packages("GGally")
install.packages("patchwork")
library(readr)
library(ggpicrust2)
library(tibble)
library(tidyverse)
library(ggprism)
library(patchwork)
library(GGally)
이후 분석이 완료된 picrust2 결과와 예제데이터의 metadata를 불러옵니다.
메타데이터를 ggpicrust2의 형식으로 변환해야 합니다.
metadata <-
read_delim(
"./meta.tsv",
delim = "\t",
escape_double = FALSE,
trim_ws = TRUE
)
metadata
# # A tibble: 34 × 9
# ...1 barcode.sequence body.site year month day subject reported.antibiotic.usage days.since.experiment…¹
# <chr> <chr> <chr> <dbl> <dbl> <dbl> <chr> <chr> <dbl>
# 1 L1S105 AGTGCGATGCGT gut 2009 3 17 subject-1 No 140
# 2 L1S140 ATGGCAGCTCTA gut 2008 10 28 subject-2 Yes 0
# 3 L1S208 CTGAGATACGCG gut 2009 1 20 subject-2 No 84
# 4 L1S257 CCGACTGAGATG gut 2009 3 17 subject-2 No 140
# 5 L1S281 CCTCTCGTGATC gut 2009 4 14 subject-2 No 168
# 6 L1S57 ACACACTATGGC gut 2009 1 20 subject-1 No 84
# 7 L1S76 ACTACGTGTGGT gut 2009 2 17 subject-1 No 112
# 8 L1S8 AGCTGACTAGTC gut 2008 10 28 subject-1 Yes 0
# 9 L2S155 ACGATGCGACCA left palm 2009 1 20 subject-1 No 84
# 10 L2S175 AGCTATCCACGA left palm 2009 2 17 subject-1 No 112
# # ℹ 24 more rows
# # ℹ abbreviated name: ¹days.since.experiment.start
# # ℹ Use `print(n = ...)` to see more rows유의할 점은 sample_name(L1 S105 등..)이 rownames로 가있는 것이 아니라, 별개의 column이어야 합니다.
이때 import 한 데이터에서 샘플 이름이 담인 열의 이름이 "... 1"로 되어있습니다. 이를 SampleID로 바꾸어 줍니다.
colnames(metadata)[1] <- "SampleID"
이 중에서 우리는 body.site가 gut와 tongue인 데이터를 추출해서 사용할 것입니다.
metadata <- metadata[metadata$body.site %in% c("gut", "tongue"), ]
picrust2 결과물도 가져와 봅니다. 아래 적힌 경로는 본인의 분석 환경에 맞게 수정해야 합니다.
kegg_abundance <-
ko2kegg_abundance(
"./pred_metagenome_unstrat.tsv"
)
kegg_abundance
"pred_metagenome_unstrat.tsv" 파일과 kegg_abundance로 import 된 파일의 행 개수가 다를 것입니다. 이는 여러 개의 ko가 하나의 kegg pathway에 속하는 경우가 있기 때문에, ko abundacne가 kegg pathway abundance로 변환되는 과정을 거치면서 테이블의 크기가 압축되는 것입니다.
그런데 ko2 kegg_abundace함수가 시행될 때마다, 결괏값이 달라지는 경우가 있습니다. 이를 대비해서 set.seed(42)를 사용해 랜덤값을 고정해 주는 것이 좋습니다..
위 결과물도 gut과 tongue에 사용된 샘플만 추출합시다.
kegg_abundance2 <- kegg_abundance[, metadata$SampleID]
2) Differential abundance (DA) 분석하기
DA란 차등발현하는 유전자/생물을 말한다. 이를 판별하게 위해 DESeq, ALDEx2등의 방법이 사용됩니다. ggpicrust2에서는 lefser, metagenomeSeq, limma_voom, edgeR, LinDA, Maaslin2, DESeq2, ALDEx2를 사용할 수 있습니다. 예제 분석은 ALDEx2를 사용해 봅시다.
ALDEx2는 input data를 Read count를 사용하며, 이를 central log ratio로 변환합니다.
반대로 Lefser(Lefse의 R버전) 같은 경우에는 relative abundance를 input으로 사용합니다.
# factor값으로 바꾸어 주기
metadata$body.site <- factor(metadata$body.site, levels = c("gut", "tongue"))
# Aldex2분석 수행
ko_AlDEx <-
pathway_daa(
abundance = kegg_abundance2,
metadata = metadata,
group = "body.site",
daa_method = "ALDEx2",
select = NULL,
reference = NULL
)
# 결과값 보기
ko_AlDEx %>% head()
# feature method group1 group2 p_values adj_method p_adjust
# 1 ko05340 ALDEx2_Welch's t test gut tongue 0.00402396 BH 0.01078215
# 2 ko00564 ALDEx2_Welch's t test gut tongue 0.17365998 BH 0.26492653
# 3 ko00680 ALDEx2_Welch's t test gut tongue 0.03217803 BH 0.06656202
# 4 ko00562 ALDEx2_Welch's t test gut tongue 0.67601305 BH 0.75152515
# 5 ko03030 ALDEx2_Welch's t test gut tongue 0.06168772 BH 0.10879943
# 6 ko00561 ALDEx2_Welch's t test gut tongue 0.02946089 BH 0.06329062
ko_AlDEx %>% tail() # 뒷부분에는 wilcoxon rank sum test결과값 위치
결과물을 보면 통계적인 분석 방법에 의한 p-value값과 BH보정이 들어간 p_adjust값을 볼 수 있습니다. 또한 두 그룹 간의 비교이기 때문에, 통계적인 분석 방법은 Welch's t test와 Wilcoxon rank test가 사용되었습니다. 마이크로바이옴 데이터는 수가 적고 모수를 추정하기 어려워서 비모수적인 통계 방법인 Wilcoxon rank test를 사용한 값만 추출해서 이후 분석에 사용해 봅시다.
ko_AlDEx_df <- ko_AlDEx[ko_AlDEx$method == "ALDEx2_Wilcoxon rank test", ]
2) Annotation 하기
KO 데이터에 대사 관련 정보를 추가해 봅시다. 이는 KO id를 KEGG와 매핑하여 온라인으로 불러옵니다. 이때 데이터의 양이 너무 많으면 에러 메시지가 뜨니 여러 번 나누어서 실행해야 합니다. 그러나 위 예제 데이터는 양이 많지 않아 한번에 돌릴 수 있습니다.
(pathway는 picrust2 결과 데이터에 따라 "EC", "KO", "MetaCyc" 중에 선택 가능)
ko_annotation <-pathway_annotation(pathway = "KO",
daa_results_df = ko_AlDEx_df,
ko_to_kegg = TRUE)※ 현재 kegg API문제로 30여 개 이상 샘플을 annotaion 할 경우 많은 시간이 걸린다. 그러므로 p-adjust top 30만 보는 것을 추천 ※
d위 글에서 ko번호로 pathway추출하여 사용하는 것을 추천드립니다.
ko_annotation %>% colnames()
[1] "feature" "method" "group1" "group2"
[5] "p_values" "adj_method" "p_adjust" "pathway_name"
[9] "pathway_description" "pathway_class" "pathway_map"이후 데이터를 확인하면 "pathway_name", "pathway_description", "pathway_class", "pathway_map" 이 추가로 생긴 것을 볼 수 있습니다.
4-1) 시각화하기: ERROR bar
Error bar는 미생물 분석 및 시각화 프로그램인 STAMP의 시각화 구조가 대표적입니다. STAMP로 그린 picrust의 예시 그림은 아래와 같습니다. 그림은 아래와 같이 왼쪽에는 해당하는 대사의 mean relative abundance의 평균값이 있고 오른쪽에는 각 그룹마다 평균값의 차이와 p-value값을 보여줍니다.
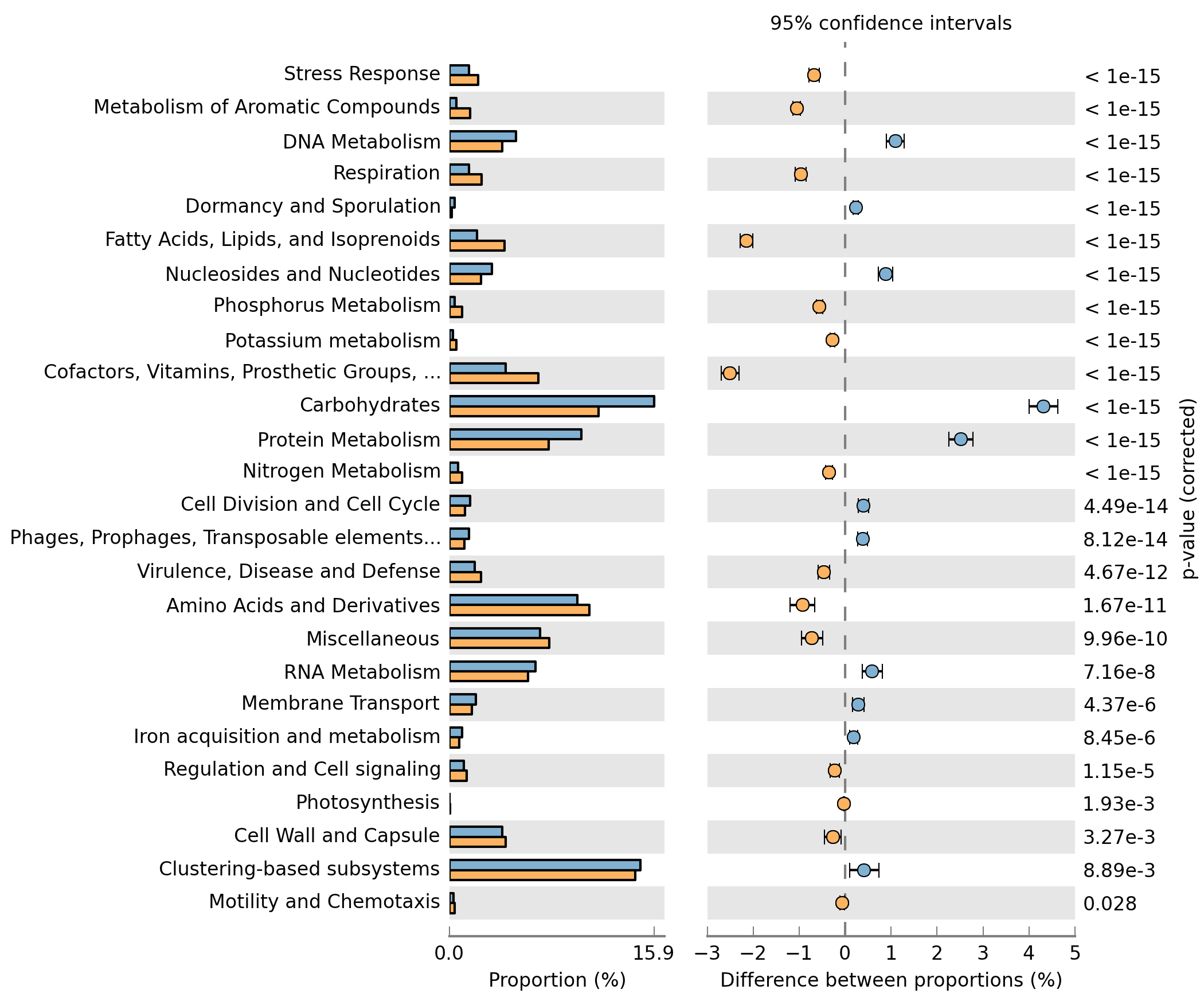
ggpicrust2에서도 동일한 그림을 그릴 수 있지만, 30개가 넘는 대사를 표현하기 어렵습니다. 그러므로 p_adjust 값이 가장 낮은 top 30을 고른 후 시각화해 봅시다.
Top30 <- ko_annotation %>% arrange(p_adjust) %>% top_n(30)
kegg_abundance_t30 <- kegg_abundance2[rownames(kegg_abundance2) %in% Top30$feature, ]
p <- pathway_errorbar(abundance = kegg_abundance_t30,
daa_results_df = Top30,
Group = metadata$body.site,
ko_to_kegg = T,
p_values_threshold = 0.05,
order = "pathway_class",
select = NULL,
p_value_bar = T,
colors = NULL,
x_lab = NULL)
p
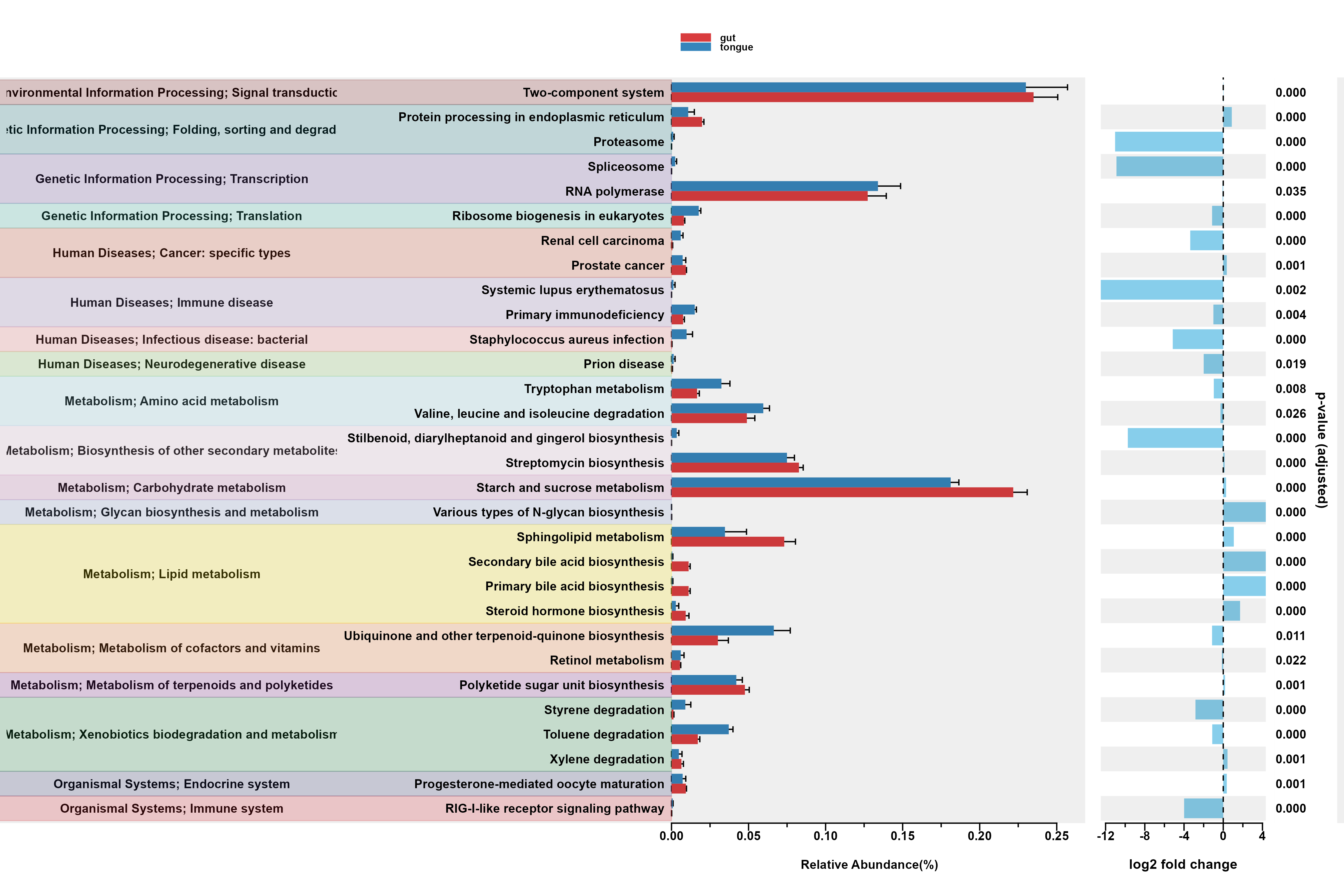
STAMP의 그림과 달리, 왼쪽에는 각 대사체의 큰 범주도 추가된 것을 볼 수 있습니다. (STAMP의 그림과는 x축의 이름이 다르지만 뜻은 동일하다.)
4-2) 시각화하기 : PCA
pca_plot <- ggpicrust2::pathway_pca(kegg_abundance_t30, metadata, "body.site")
pca_plot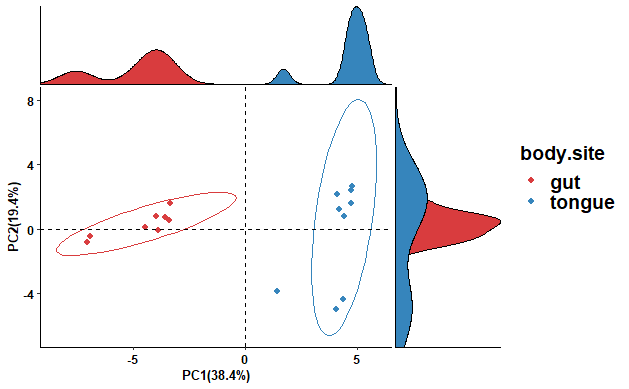
4-3) 시각화하기 : heatmap
colnames(metadata)[1] <- "sample_name"
heatmap_plot <- ggpicrust2::pathway_heatmap(kegg_abundance_t30, metadata, "body.site")
heatmap_plot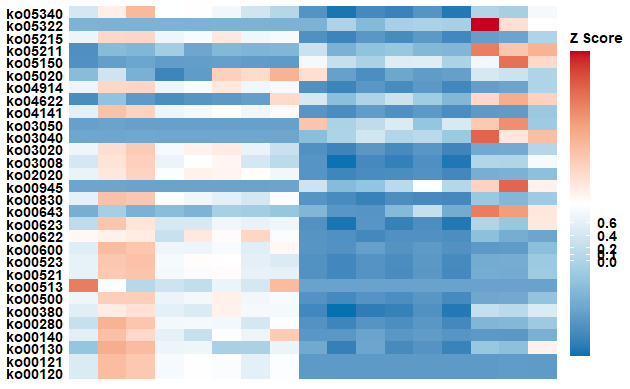
| 참고
Chen Yang, Aaron Burberry, Xuan Cao, Jiahao Mai, Fabio Cominelli, Liangliang Zhang. (2023). ggpicrust2: an R package for PICRUSt2 predicted functional profile analysis and visualization. arXiv preprint arXiv:2303.10388.
ggpicrust2: an R package for PICRUSt2 predicted functional profile analysis and visualization
Microbiome research is now moving beyond the compositional analysis of microbial taxa in a sample. Increasing evidence from large human microbiome studies suggests that functional consequences of changes in the intestinal microbiome may provide more power
arxiv.org
Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J, Knights D, Gajer P, Ravel J, Fierer N, Gordon JI, Knight R. Moving pictures of the human microbiome. Genome Biol. 2011;12(5):R50. doi: 10.1186/gb-2011-12-5-r50. PMID: 21624126; PMCID: PMC3271711.
Moving pictures of the human microbiome - PubMed
DNA sequencing and computational advances described here provide the ability to go beyond infrequent snapshots of our human-associated microbial ecology to high-resolution assessments of temporal variations over protracted periods, within and between body
pubmed.ncbi.nlm.nih.gov
| 오류 문의
pathway_annotation()을 시행했을 때, 기존의 input 파일로 사용된 데이터보다 output의 데이터 크기가 손실되었다.
pathway_annotation은 p-value값이 0.05 이하인 값만 결과만 내놓는다. 그러나 0.05 값 이하의 데이터들에서도 일부 손실이 발생하였다. 이를 패키지 제작자에게 문의하였으며, 답변을 얻었다.
이는 ggpicurst2 패키지의 한계가 아니라 KEGG에서 ko id에 맞는 정보를 다운로드하는 API의 자체적인 문제라고 한다. 온라인 데이터베이스 KEGG에서 정보를 가져올 때는 R의 keggGet을 사용한다. 위 패키지를 사용하면 심심치 않게 오류가 생기는 것을 볼 수 있다. 이를 극복하기 위해, ggpicrust2의 제작자는 가능한 작은 단위로 데이터를 나누로, kegg의 과부하가 걸리지 않게 일종의 휴식기간을 주면서 데이터를 다운로드하여야 한다고 한다고 답변했다. 추가로 전달받은 코드는 아래와 같다.
ko_id # kegg 에 검색하고자 하는 ko id
# Split the dataframe into subdataframes every 20 rows_20개씩 나누기
split_data <- split(ko_id, ceiling(seq_along(ko_id)/20))
# Initialize an empty result list_keggGet의 결과가 저장되는 리스트 만들기
KeggGet_out <- list()
# Use a for loop instead of lapply to add a pause between iterations_반복하면서 시행
for (i in seq_along(split_data)) {
kos <- split_data[[i]]
for (j in kos) {
result <- tryCatch(keggGet(j), error=function(e) NULL)
KeggGet_out[[i]] <- result
Sys.sleep(60) # API의 과부화를 막기 위해 한 데이터 셋을 다운 받고 1분의 휴식 시간을 가진다
}
}
# Combine all results into one data frame_결과 합치기(list -> data.frame)
final_result <- bind_rows(KeggGet_out)API의 과부하를 막기 위해 휴식시간을 주었기 때문에 다운로드 시간은 매우 길어진다(약 7000개 데이터를 실행하는 데에 1~2일이 걸렸다). 또한 가운데 tryCatch(keggGet(j), error=function(e) NULL) 부분은 직접적으로 kegg 데이터를 가져오는 함수이며, 이를 ggpicrust2::pathway_annotation()로 바꾸어도 상관없다.
직접 제작자에게 문의해 본 일은 이번으로 두 번째이다. 처음 질문은 답변조차 받지 못했다. 그런데 ggpicrust2 제작자인 cafferychen777은 문의한 당일, 그것도 한 시간 안되어서 답변을 적어주었다. 또한 끝인사로 사용해 줘서 감사하고 언제든지 문의를 달라고 했으며, 나의 성공을 빌어주었다. 정말 친절하고 멋진 개발자였다.
또한 이 분이 하고 있는 프로젝트로는 마이크로바이옴 시각화 자료를 공유하는 갤러리가 있다. 아직 공사 중이지만, 내가 하고자 했던 목적(정보의 공유)과 일치하는 프로젝트임으로, 완성된다면 열심히 참여하리라고 생각했다.